

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Paratuberculosis (PTB) or Johne's disease is a chronic and debilitating disease in ruminants that is caused by Mycobacterium avium subsp. paratuberculosis (MAP). The disease is characterized by intermittent to persistent diarrhea, progressive wasting, and eventually leads to death. Currently, there are no treatments available to cure the infection [5]. The disease occurs worldwide and causes serious economic losses to the cattle industry due to premature culling and production losses [27]. In most of the major dairy-producing countries, it has been estimated that more than 50% of dairy cattle herds were infected with MAP [1819]. In Korea, the national prevalence of PTB in individual cattle was estimated at 3.3% to 7.1% until 2010 [31]. Recently, 12 of 37 herds (32.4%) tested in Korea were found to be MAP-positive, as determined by performing polymerase chain reaction (PCR) of fecal DNA samples [20]. Despite the high herd-level prevalence, there have been no reports on the molecular characterization of MAP strains isolated from cattle in Korea. Understanding the genetic diversity of MAP is necessary for establishing disease control strategies [7]. Molecular typing methods for MAP strains provide important information for genetic diversity and evolutionary relationships and can assist in elucidating inter- and intra-species transmissions [21426]. Several methods for subtyping of MAP have been developed. PCR and restriction endonuclease analysis (PCR-REA) of the IS1311 locus of MAP has been used, and it allows for the differentiation of MAP linages into three subtypes (cattle-, sheep-, and bison-type) according to host preferences [2230]. Recently, typing techniques such as variable-number tandem repeats of mycobacterial interspersed repetitive units (MIRU-VNTR) and multilocus short sequence repeat (MLSSR) have been used to differentiate subtypes [26781122262829]. The techniques using these targets provide high discriminatory power and are easy to use [228]. The aim of this study was to describe the genetic diversity of MAP isolated from individual cows in Korea by using IS1311 PCR-REA, MIRU-VNTR, and MLSSR typing methods.
Materials and Methods
Collection of fecal DNA positive in MAP
During 2013 and 2014, detection of MAP by PCR was conducted by using fecal material collected from 37 herds across 6 provinces of Korea [20]. From a total of 1,562 fecal DNA samples, 35 were shown to be positive for MAP IS900 and ISMap02. The IS1311 targeting PCR was conducted using fecal DNA, and the PCR-positive fecal DNA samples were subjected to subsequent REA analysis.
Bacterial isolation
Nineteen Korean MAP isolates and 8 foreign isolates were used for genotyping. Korean isolates were obtained from feces or tissues that were collected from 5 herds in 3 provinces (Chungnam, Jeonbuk, and Jeju). Among them, 6 isolates were obtained from 35 of fecal PCR-positive fecal samples. Along with the Korean isolates, three foreign MAP strains kindly provided by Dr. Jacek Gwozdz (Johne's Disease Laboratory, Department of Primary Industries, Australia) and 5 DNA samples kindly provided by Dr. Iva Slana (Veterinary Research Institute, Czech Republic) were used in this study to compare their epidemiological characteristics with those of the Korean isolates. The culture of MAP from feces and tissues was conducted by using the VersaTREK system (Thermo Scientific, USA). Briefly, fecal samples were processed for decontamination by using a modified Cornell method as described previously [4]. Tissue decontamination was conducted as described previously [17]. After decontamination, the suspension was inoculated into VersaTREK Para-JEM broth (Thermo Scientific) containing para-JEM supplements (para-JEM GS, para-JEM AS, para-JEM EYS, and para-JEM blue [Thermo Scientific]) and then incubated at 37℃ in the VersaTREK instrument (Trek Diagnostic System; Thermo Scientific). After incubation for six weeks, tubes that were shown by the instrument to have a positive result were inoculated into modified Herrold's egg yolk medium with mycobactin J (IDvet, France) and cultured for six weeks. The sub-cultured colonies were confirmed positive by using MAP species-specific primers such as IS900 and ISMap02 in duplex real-time PCR.
DNA preparation of MAP isolates
A colony of each isolate was cultured in 5 mL of Middlebrook 7H9 broth supplemented with Middlebrook OADC and mycobactin J (2 mg/L). After incubation at 37℃ for 6 weeks, the bacterial suspension was centrifuged at 3,000 × g for 20 min, and the pellet was lysed in 1 mL of L6 lysis buffer (5.25 M GuSCN, 50 mM Tris-HCl at pH 6.4, 20 mM EDTA, 1.3% Triton X-100, distilled water). Subsequent processes were carried out as previously described [21].
Characterization of MAP isolates/fecal DNA with IS1311 PCR-REA
Polymorphism present in 223 bp of IS1311 was analyzed by performing IS1311 PCR-REA to characterize the three types (cattle-, bison-, and sheep-type) of MAP isolates and fecal DNA samples. The IS1311 PCR was conducted by using M56 and M119 primers following the method of Marsh et al. [15] with some modifications. The PCR products that were 608 bp were then digested by HinfI [22]. The band patterns were visualized after electrophoresis on a 2% agarose gel.
Genotyping of MAP isolates with MIRU-VNTR
Genotyping of the 27 MAP isolates was conducted by using eight MIRU-VNTR targets (292, X3, 25, 47, 3, 7, 10, and 32) that were established previously [28]. PCR amplification of each locus was conducted following amplification conditions described previously [28]. The repeat numbers of the PCR products were analyzed after electrophoresis on 2% agarose gel. The PCR results and MAP strains were identified according to information in the Mac INMV database.
MLSSR analysis with MAP isolates
The MLSSR analysis was carried out by performing PCR amplification and sequencing. Eleven short sequence repeat (SSR) loci were analyzed as described previously [2]. The PCR amplicons were then sequenced by the DNA sequencing service (Macrogen, Korea), and the number of SSRs in each locus was identified.
Calculation of discriminatory power
The discriminatory index (DI), as described by Hunter and Gaston [10], was used as a numerical indicator of the discriminatory power of the MIRU-VNTR and MLSSR typing methods. The DI was calculated using the following equation:
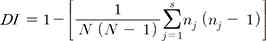
where N is the total number of isolates in the typing scheme, s is the total number of distinct types discriminated by each typing method, and nj is the number of isolates belonging to the jth type.
Genetic relationship analysis
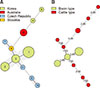
The genetic relationships among the 27 strains were investigated by creating minimum spanning trees (MST) using 11 SSR markers. The MST was created by using BioNumerics software (Applied Maths, USA). Through the MST, the shortest possible distance between nodes was linked.
Results
IS1311 PCR-REA typing
Eighteen of 35 fecal DNA samples showed the expected sizes in the IS1311 PCR results. Among the 18, six were identical to MAP isolates cultured from the fecal samples. The remaining 12 fecal DNA samples were analyzed by IS1311 PCR-REA typing. Among the 31 samples (12 fecal DNA and 19 MAP isolate samples) obtained from Korea, 8 samples had band patterns representative of cattle-type genotypes, while 23 had bison-type genotype patterns (Fig. 1, Table 1). All 8 foreign MAP isolates had cattle-type genotype patterns. Two herds each located in Korean regions 3 and 6 showed both genotypes, while other herds only showed one genotype. The cattle-type strain was detected in 6 herds located in 5 regions in 3 Korean provinces, while the bison-type strain was detected in 7 herds located in 5 regions in 4 Korean provinces (Table 1).
MIRU-VNTR typing
Genotyping using the MIRU-VNTR method differentiated 4 subtypes among the 27 MAP isolates (Table 2). The most prevalent type was INMV 68, which was present in 63.0% (n = 17) of the isolates. The INMV 2, INMV 1, and INMV 5 types were present in 18.5%, 14.8%, and 3.7% of the isolates, respectively. Three MIRU-VNTR loci (MIRU 292, VNTR 25, and VNTR 10) were polymorphic. Three different alleles were observed in MIRU 292, whereas only two variable alleles were observed in VNTR 25 and VNTR 10. Among the domestic isolates, two MIRU-VNTR types (INMV 2 and INMV 68) were observed (Table 3). Two cattle-type strains isolated from a herd were identified as INMV 2, whereas all isolates identified as bison-type were from five herds in three regions belonged to the INMV 68 type. Foreign isolates identified as cattle-type were differentiated into three INMV types according to the country in which they were isolated. All three isolates from Australia were identified as INMV 2 subtypes, whereas four isolates from the Czech Republic were identified as INMV 1, and an isolate from Slovakia was INMV 5. Allelic diversity was calculated as 0.567 when both domestic and foreign isolates were included in the calculation. The DI value for domestic isolates only was 0.199. The MIRU 292 showed the highest allele diversity observed in this study. The allelic diversities for each locus were 0.556 for MIRU 292, 0.484 for VNTR 25, and 0.074 for VNTR 10.
MLSSR typing
Eleven SSR loci were characterized in 27 MAP isolates. The MLSSR analysis identified 11 subtypes among the 27 isolates (Table 4). The most prevalent type was MLSSR type 1, which was identified in 8 isolates (29.6%). Of the 11 MLSSR types detected in this study, 7 were unique to one isolate (3.7%). Among the 11 MLSSR loci, loci 1, 2, 6, 7, 8, and 9 were polymorphic with two to four different alleles, whereas the others did not show any polymorphisms. Locus 2 had the highest allelic diversity with a DI value of 0.707. The allelic diversity of loci 1, 6, 7, 8, and 9 were 0.074, 0.313, 0.544, 0.313, and 0.262, respectively. The overall DI value of the MLSSR typing was calculated to be 0.866. Among the domestic isolates, five MLSSR types were detected and the DI for those isolates was 0.743 (Table 3). Five isolates from the Czech Republic and Slovakia showed distinctive MLSSR types, whereas three Australian isolates had the same MLSSR type (Table 3). There were no MAP strains that had similar MLSSR types among the three different countries assessed in this study.
Genetic relationship
The genetic relationship between the various strains was constructed by creating MST (Fig. 2). The 11 SSR loci were used for MST construction among the 27 isolates. In Fig. 2, the size of each node indicates the population size of the sample, and the country of origin is labeled with their respective color. The predominant genotypes were types 1, 2 and 3, which all belong to the bison-type genotype. In addition, the strains corresponding to INMV 1 and INMV 5, which were isolated in the Czech Republic and Slovakia, were clustered in the same group with the bison-type strains. MLSSR types 4, 5, and 6, which belonged to INMV 2, were not clustered in the same group because the clonal distance between these strains was calculated as 2.0.
Discussion
In the present study, three molecular typing methods, IS1311 PCR-REA, MIRU-VNTR, and MLSSR were applied in an epidemiological analysis of MAP isolated in Korea. The IS1311 PCR-REA results showed that cattle-type and bison-type genotypes co-existed in Korean cattle farms, and especially, the bison-type strains were distributed nationwide. Because bison-type strains are not common in cattle in other countries [24], it is thought that the wide distribution of the bison-type in Korea has an important meaning in an epidemiological aspect. Bison-type strains of MAP have been reported in several countries other than Korea [13232425]. On the American continent, Sohal et al. [24] reported bison-type strains isolated from domestic species in Quebec, Canada. In India, the bison-type strain was the predominant strain identified in most domestic ruminant species, and that type was also found in some wild animals [2325]. In Korea, bison-type strains were reported in wild boars and domestic species such as cattle, goat, and elk [13]. Because the bison-type genotype was discovered in both wildlife and domestic animals in Korea, it could be suggested that wildlife species have the potential to spread MAP between wildlife and domestic cattle. However, detailed epidemiological studies are needed to clarify the relationship between two host species.
Through the MIRU-VNTR analysis, four INMV types were observed in domestic and foreign isolates. The characteristic feature of the MIRU-VNTR genotyping was that all 17 bison-type isolates appeared as the INMV 68 type. The reason for the low DI of the MIRU-VNTR value may be because many isolates belonged to one group. The INMV 68 type has been reported previously in other bison-type strains in Canada [124]. Therefore, the results may suggest that MIRU-VNTR loci are highly conserved in bison-type MAP strains. Two cattle-type domestic isolates were identified as INMV 2, while other foreign isolates were identified as INMV 1 or INMV 2, except for 1 isolate identified as INMV 5 from Slovakia. The INMV 1 and INMV 2 are the most prevalent types observed worldwide [1691625], and the INMV 5 type has been reported in Argentina and Germany [811]. In this study, no unique INMV types were observed in the MIRU-VNTR genotyping results. Although two common INMV types were observed in Korea, that result does not mean that there were only two introductory events in Korea because MIRU-VNTR may overestimate relatedness [1]. Ahlstrom et al. [1] pointed out the limitations of the MIRU-VNTR method by identifying that several isolates belonging to three common INMV types were highly unrelated to each other through a single nucleotide polymorphism analysis. Therefore, an additional typing method with a high discriminatory power was needed to obtain more detailed information about the epidemiological status of MAP in Korea.
The MLSSR molecular typing discriminated 27 isolates into 11 subtypes, especially as bison-type isolates, that were not discriminated by MIRU-VNTR; those subtypes were divided into three different MLSSR types. The SSR loci 3, 4, 5, 10, and 11 were not variable for MAP isolates as has been reported in other studies [2729]. The most variable SSR marker for the isolates was locus 2, which revealed four different alleles. Several previous studies have reported that the combined results of MIRU-VNTR and MLSSR typing produce the highest DI values [7824]. In this study, however, there were no MLSSR types that were divided into different INMV types, suggesting that the MLSSR alone has sufficient discriminatory power in cattle-derived MAP strains. However, there is a potential disadvantage of MLSSR because a high number of G repeats at loci 1 and 2 may not be accurately read due to a sequencing technology limitation [7]. Therefore, sequencing of loci 1 and 2 was performed repeatedly in both directions to increase the reliability of the sequencing results. In addition, all alleles > 11 were classified into the same type in this study, as also described in previous studies, thereby limiting the interpretation [729]. Another potential disadvantage of the MLSSR typing is that the SSR locus 2 has questionable stability. There is a claim that the SSR locus 2 is unstable for epidemiological study because strand slippage events could occur during chromosomal duplication in the course of bacterial spreading [12]. In that study, different G repeat numbers at SSR locus 2 were observed in multiple isolates from individual animals or from a single cattle herd. This instability argument could be supported indirectly by the present study because the bison-type strains were divided into three subtypes only by the SSR locus 2, and all of the subtypes were detected in one herd. However, the study reported by Kasnitz et al. [12] showed that the SSR locus 2 was stable after 12-fold subculture in vitro. Therefore, the three different bison-type strains identified in this study need to be further validated through detailed genotyping methods such as whole genome sequencing (WGS)-based analysis [314].
The clonal distance of each strain obtained by using the MLSSR typing results are shown as an MST. The characteristic features of the MST were that the cattle-type and the bison-type did not appear as two distinct clusters. Instead, the INMV 1 and 5 types were clustered to the same group as the bison-type strains. It has been reported that INMV 1 strains are closely related to the bison-type based on an MST analysis using the combination of MIRU-VNTR and MLSSR markers [24]; however, the relationship between INMV 5 and the bison-type genotype has not yet been clarified. Based on these results, it can be inferred that the bison-type is not phylogenetically distinguished from the cattle-type. Recently, type B (bison-type) strains were classified as a subgroup of type C (cattle-type) through WGS-based genotyping [3].
A limitation of this study is that the results of the analysis cannot represent the whole Korean epidemiological situation because the number of MAP isolates/DNA samples was low. However, the samples used in this study, especially the MAP-positive fecal DNA samples that were obtained from several herds nationwide and used in the prevalence test, could reflect the present situation in Korea to a certain extent. To the best of our knowledge, this is the first epidemiological survey report about MAP strains isolated from cattle in Korea that used both MIRU-VNTR and MLSSR typing methods.
In conclusion, MLSSR typing is a highly discriminatory genotyping method that can be used as an alternative to MIRU-VNTR genotyping. Moreover, the genotyping results in this study indicate that the prevalent MAP strains in cattle in Korea may be bison-type strains, and this finding has important epidemiological meaning for the control and prevention of PTB in Korea.





 XML Download
XML Download