

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Heart disease remains the leading cause of death worldwide. The heart has a limited capacity for regeneration and repair. Cell death is a hallmark of various heart diseases, including heart failure (HF), myocardial infarction (MI), and ischemia/reperfusion (I/R).1) Cardiac myocytes express Fas cell surface death receptor (FAS) and tumor necrosis factor (TNF) receptor 1, which have both been implicated in cardiovascular pathology through stimulation of FAS ligand, TNF-α, or TNF-related apoptosis-inducing ligand (TRAIL).1) TRAIL, a member of the TNF superfamily, is expressed either as a transmembrane protein on the cell surface of a variety of cell types or is released as a soluble protein.2) In humans, four membrane-bound TRAIL receptors have been identified. Among these, TRAIL-R1 (death receptor [DR]-4) and TRAIL-R2 (DR5) are apoptosis-inducing receptors, and TRAIL-R3 (decoy receptor [DcR] 1) and TRAIL-R4 (DcR2) act as decoy receptors because of absent or nonfunctional death domains.3) TRAIL binds to the death receptors DR4 and DR5, and triggers the apoptosis signaling pathway by recruiting Fas-associated death domain (FADD), subsequently activating caspase-8. Caspase-8 activates executioner caspases, such as caspase-3, -6, and -7, leading to cleavage of numerous intracellular proteins.4) Unlike FasL and TNF-α, TRAIL selectively induces cell death in malignant cells but not in normal cells5) because of the relative selectivity of DR4 and DR5 for malignant cells over normal or healthy cells.6)7) However, up-regulation of DR4 or DR5 in normal cells can induce apoptosis by stimulating TRAIL. Numerous signaling molecules trigger DR5 and DR4 induction, including activation of mitogen-activated protein kinases (MAPKs) and the binding of the CCAAT/enhancer binding protein (C/EBP) homologous protein (CHOP) to the DR5 promoter.8)
β-Adrenergic receptors (β-AR) play key roles in the regulation of cardiac function, both under normal conditions and under pathological conditions, such as during HF.9) Chronic impairment of β-AR signaling contributes to alterations in cardiac structure through increased apoptosis, hypertrophy, fibrosis and steadily decreased contractile function as HF progresses.10)11) Isoproterenol (ISO), an agonist of the adrenergic receptor, induces compromised and unstable cardiac hemodynamics and aggravates cell death, thereby inducing HF.12)13) Several related signaling pathways including those involved in oxidative stress, calcium dysregulation and activation of apoptotic genes contribute to ISO-induced heart damage. Apoptosis is a critical step in provoking systolic dysfunction and congestive HF.14) Clinical studies have demonstrated that adrenergic blockers significantly improve cardiac function and reduce cardiac apoptosis in patients with HF.15) However, the mechanism of ISO action in TRAIL-induced cardiac apoptosis remains unknown.
Therefore, we investigated whether ISO enhanced TRAIL-induced apoptosis in human embryonic kidney (HEK) 293 cells. In this study, we report that TRAIL-induced apoptosis is synergistically increased by ISO through the up-regulation of DR5.
Materials and Methods
Cell culture
Human embryonic kidney (HEK 293) cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Rockville, MD, USA) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO, USA), streptomycin (50 µg/mL) and penicillin (100 U/mL). Cells were incubated at 37℃ in a humidified atmosphere of 5% CO2.
Viability assay
HEK 293 cells were plated at 1×104 cells/cm2 in 96-well plates. After 24 hours, the medium was changed to LG-DMEM, and ISO (100 µM) and/or TRAIL (100 ng/mL) were added. Cells were cultured in each condition for an additional 24 hours. Then, 0.5 mg of methylthiazolyldiphenyl-tetrazolium bromide (MTT: Sigma-Aldrich, San Diego, CA, USA) dissolved in phosphate-buffered saline (PBS) was added to each well (final concentration, 5 mg/mL),16) and cells were incubated at 37℃ for 1 hour. MTT formazan that formed was dissolved in 100 µL dimethylsulfoxide and incubation was continued for an additional 15 minutes with stirring before the optical density of each well at 570 nm was determined using a microplate reader.
Immunoblotting
HEK 293 cells were treated with ISO (100 µM) and/or TRAIL (100 ng/mL) for 24 hours. Cells were harvested, washed with ice-cold PBS and solubilized using lysis buffer (0.1% sodium dodecyl sulfate [SDS], 1% Triton X-100, 50 mM Tris-HCl [pH 7.4], 150 mM NaCl) containing Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific, Rockford, IL, USA). Proteins were separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to an Immobilon membrane. Each membrane was blocked with 5% skim milk in TBST (Tris-HCl buffered saline containing 0.1% Tween 20) and incubated with primary antibodies against caspase (Cas)-3, cleaved Cas-3, and poly (ADP-ribose) polymerase (PARP; 1:1000; Cell Signaling Technology, Danvers, MA, USA), DR5 (1:1000, R&D Systems, Minneapolis, MN, USA) and glyceraldehydes-3-phosphate dehydrogenase (GAPDH; 1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Bound primary antibodies were detected with horseradish peroxidase-conjugated secondary antibody (1:2000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), treated with EZ-Western Lumi Pico (DOGEN, Seoul, Korea) and visualized using the FluorChem FC2 system (Alpha Innotech, Santa Clara, CA, USA).
Fluorescein isothiocynate-annexin-V/propidium iodide staining
A fluorescein isothiocynate (FITC)-annexin-V apoptosis detection kit (BD Biosciences, Franklin Lakes, NJ, USA) was used according to the manufacturer's instructions. Cells were harvested, washed twice with cold PBS and re-suspended in 1xbinding buffer. Cells were stained with FITC-annexin-V and PI for 15 minutes at room temperature in the dark. Cells were analyzed on a flow cytometer without washing within 1 hour of the completion of staining.
Results
Cell viability by isoproterenol and/or tumor necrosis factor-related apoptosis-inducing ligand
To investigate whether ISO can enhance TRAIL-induced apoptosis, HEK 293 cells were treated with ISO (100 µM) and TRAIL (100 ng/mL) for 24 hours. Cell viability of the treated cells was examined by light microscopy, and cytotoxicity of ISO and/or TRAIL was quantified using the MTT assay (Fig. 1). ISO or TRAIL treatment decreased the viability of HEK 293 cell populations by 13% and 17%, respectively (Fig. 1B). However, co-treatment with ISO and TRAIL resulted in the cell death of 35% of HEK 293 cells after 24 hours. This result suggested that ISO enhances TRAIL-induced apoptosis in HEK 293 cells.
Cas-3-dependent apoptosis resulting from co-treatment with isoproterenol and tumor necrosis factor-related apoptosis-inducing ligand
To further confirm that co-treatment with ISO and TRAIL is cytotoxic, apoptosis in HEK 293 cells was evaluated by flow cytometry using annexin-V and PI. Increases were apparent in early apoptotic cells (i.e., annexin-V positive/PI negative; 19.4%), late apoptotic cells (i.e., annexin-V positive/PI positive; 6.3%) and dead cells (i.e., annexin-V negative/PI positive; 1.1%) when cells were co-treated with ISO and TRAIL, compared to cells treated with ISO or TRAIL (Fig. 2A and B). Next, caspase-3 activation and PARP degradation were examined using an immunoblotting assay. Both events are commonly observed in apoptotic cell death. Precursor Cas-3 (32 kDa) can be cleaved into 17 kDa and 12 kDa fragments, resulting in activation. PARP can be cleaved by caspases, which separates the PARP amino-terminal DNA binding domain (24 kDa) from the carboxy-terminal catalytic domain (89 kDa) in response to apoptotic signals. Substantial amounts of the 17 kDa cleaved cas-3 and 89 kDa cleaved PARP were observed in HEK 293 cell preparations co-treated with ISO and TRAIL (Fig. 2C and D). These results suggested that ISO and TRAIL synergistically induce apoptosis in HEK 293 cells.
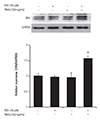
Up-regulation of death receptor 5 by co-treatment with isoproterenol and tumor necrosis factor-related apoptosis-inducing ligand
TRAIL (1 mg/mL) increases the level of DR5, but not DR4, in HEK 293 cells.17) We investigated whether DR5 could be up-regulated by co-treatment with ISO and TRAIL in HEK 293 cells. The cells were treated with ISO (100 µM) and/or TRAIL (100 ng/mL) for 24 hours. The expression of DR5 protein increased in cells co-treated with ISO and TRAIL, although individual treatment with ISO or TRAIL did not induce increased DR5 expression (Fig. 3). Taken together, the results indicated that co-treatment with ISO with TRAIL may be responsible for death of HEK 293 cells through DR5 up-regulation.
Discussion
We report that co-treatment with ISO and TRAIL enhances HEK 293 cell death through DR5 up-regulation, although individual treatment with either ISO or TRAIL does not increase DR5 expression. This implies an important finding with respect to understanding the mechanism of cell death induced by β-AR activation. Contrary to other TNF-family members, membrane-bound TRAIL is conditionally expressed in immune cells, such as natural killer cells, B cells, monocytes and dendritic cells following cytokine stimulation.18)19)20)21) Intracellular stores of TRAIL have also been found in polymorphonuclear neutrophils22)23) and are released in response to a variety of stimuli.24)25) Moreover, β-AR activation induces inflammatory responses in the heart; in vivo ISO infusion activates nuclear factor-κB (NF-kB), κB-responsive TNF-α, and interleukin-1β, and -6 in the heart.17)26) TRAIL is released by cardiac myocytes,27) but no additional information is available about the effect of TRAIL on the heart. Taken together, the prior and current results suggest that inflammation induced by β-AR activation may be responsible for TRAIL expression in cardiac myocytes and activated immune cells. Subsequently, β-AR activation and TRAIL expression induce cardiac cell death through DR5 up-regulation, which promotes apoptosis.
Various mechanisms of DRs up-regulation have been reported.28) Briefly, production of reactive oxygen species (ROS), expression of CCAAT/enhancer-binding protein -homologous protein, p53, Sp1 and Yin Yang 1 (YY1), and activation of extracellular signal-regulated kinase, c-jun N-terminal kinase and NF-κB regulate expression of DRs.28) In particular, TRAIL induces DR5 expression in several cell types. The TNF family including TRAIL induces ROS production.29)30) ROS regulate DRs through modulation of various proteins including p53.28) In addition, TRAIL (1 mg/mL) increases expression of DR5 in HEK 293, MCF-7 and MDB-MB-231 epithelial cell lines through NF-kB activation, without an effect on DR4 expression. Blockage of NF-κB activation, either by expression of dominant negative I-κB or treatment with the proteasome inhibitor lactacystin, eliminates TRAIL-induced DR5 expression.17) Moreover, by overexpressing the p65 subunit of NF-κB, which increases NF-κB transcriptional activity, DR5 expression is increased compared to vector-only-expressing cells.17) Thus, TRAIL-mediated NF-κB activation increases DR5 expression, thereby amplifying the apoptotic response of TRAIL in epithelial derived cells.17)
Although we did not analyze the mechanisms through which expression of DR5 is increased, treatment with only TRAIL did not induce DR5 expression, while co-treatment with ISO and TRAIL increased DR5 expression in HEK 293 cells. Taken together, our results suggest that treatments combining ISO with TRAIL may be responsible for HEK 293 cell death through DR5 up-regulation. More detailed studies of cardiac cell death conditions, such as β-AR activation and inflammation in the heart, will be required to understand the pathogenesis of cardiac disease.


 XML Download
XML Download