

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Conditions leading to sinus node dysfunction have considerable clinical importance. Sick sinus syndrome (SSS) manifesting as sinus bradycardia, sinus arrest, and/or sinoatrial block is the most frequent worldwide clinical indication for pacemaker implantation.1) Benson et al.2) screened SCN5A as a candidate gene in ten patients from families diagnosed with congenital SSS on the basis of disorders of cardiac rhythm and conduction during the first decade of life. Moreover, several investigations have linked variations in SCN5A, the gene encoding the pore-forming α-subunit of the cardiac-type sodium ion (Na+) channel, Nav1.5, with familial SSS.3)4)5) Furthermore, the SCN5A gene is known to be associated with different forms of arrhythmias such as long QT syndrome, Brugada syndrome, progressive cardiac conduction defect, atrial fibrillation, dilated cardiomyopathy, and overlapping syndromes.6)7)
Voltage-gated Na+ channels are transmembrane proteins that produce the fast inward Na+ current responsible for the depolarization phase of the cardiac action potential. Inherited variations in SCN5A result in a spectrum of disease entities termed as Na+ channelopathies.
To date, although many variations of the SCN5A gene have been documented in various cardiac diseases, SCN5A variations in Korean SSS patients have not yet been studied in detail. Therefore, we carried out complete sequencing of the coding regions of the SCN5A gene, excluding untranslated regions, in Korean SSS patients in order to identify any potential variations associated with SSS.
Subjects and Methods
Patient population
This study was approved by the Ethics Committee at each hospital; consent was obtained from all individuals before enrollment into the study. We enrolled 30 Korean patients with SSS and 80 controls with no cardiac symptoms. The diagnostic criterion for SSS was the presence of a pause longer than 3 seconds in the day-time during Holter monitoring or surface electrocardiography. Normal sinus function was confirmed in controls through a cardiac electrophysiological study after ablation for supraventricular tachycardia. We excluded the patients with atrial and ventricular arrhythmia such as atrial and ventricular tachycardia, atrial fibrillation and atrial flutter, atrioventricular block, Brugada syndrome, known malignancy, and a structurally abnormal heart.
Sampling and DNA extraction
All patient and control samples were recruited from the four participating medical centers: Keimyung University, Yeungnam University, Catholic University of Daegu, and Daegu Fatima Hospital. Peripheral bloods were collected in ethylenediamine tetraacetic acid (EDTA) containing tubes, and DNA was extracted from whole blood samples using the QIAamp DNA blood mini kit (Qiagen, Hilden, Germany). DNA concentration was determined using a NanoDrop ND1000 spectrophotometer, and the purity of the DNA was assessed based on the 260/280 nm absorbance ratio.
Multiplex polymerase chain reaction and sequence analysis
The coding region (exon 2-exon 28) of SCN5A was amplified by multiplex polymerase chain reaction (PCR) using newly designed primers (Table 1). PCR conditions were as follows: after an initial denaturation at 95℃ for 15 min, denaturation at 94℃ for 30 sec, annealing at 68-70℃ for 30-60 sec, and extension at 72℃ for 60-90 sec were repeated for 30-35 cycles. Following multiplex PCR, the reaction mixture was electrophoresed in a 2% agarose gel and stained with ethidium bromide (EtBr). Amplified PCR products were purified using the QIAquick PCR purification kit (Qiagen, Hilden, Germany), and directly sequenced using the BigDye Terminator Ver 3.1 cycle sequencing kit (Applied Biosystems, Foster city, CA, USA) and an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Sequencing results were compared with reference sequences (SCN5A/NM_198056.2/ENSG00000183873/ENST00000333535) using the alignment program BLAST 2.0 of the national center of biotechnology information (NCBI; Bethesda, MD, USA), and the portion of variation that occurred was determined.
Three-dimensional modeling of SCN5A
The structural model of the human SCN5A domain was obtained from the Automated SWISS-MODEL version 8.05 (Swiss Institute of Bioinformatics Biozentrum, Basel, Switzerland).8)9)10) Superimposition, model building, construction of insertion regions, structure validation, and calculation of structural properties were carried out using the subprograms ProMod, SPDBV, Loop, LoopDB, Parameters, and Topologies, which are available in the Automated Swiss-Model Package Program (http://swissmodel.expasy.org). Pymol v0.99 and Cn3D (www.ncbl.nlm.nih.gov/Structure/CN3D/cn3d.html) were used to display the three-dimensional model structures.
Patch-clamp recording
Whole-cell configuration of the voltage-clamp technique was used as described elsewhere.11) Briefly, whole-cell configuration was made in Tyrode's solution. Pipette resistances were 1.5-3 MΩ. After achieving a gigaseal, the test-pulse current was nulled by adjusting the pipette capacitance compensator with both fast and slow components. After break-in, the whole-cell charging transient was nulled by adjusting whole-cell capacitance and series resistance. Voltage control protocols were generated with an Axopatch 200B amplifier/Digidata 1440A acquisition system using the pCLAMP-10 software (Molecular Devices/Axon, Sunnyvale, CA, USA). Whole-cell recording was analyzed using Clampfit 10.2 (Axon, Sunnyvale, CA, USA). For measuring INa, we used Tyrode's solution (see above) as the bath solution. The pipette solution contained (in mM) NaF 10, CsF 110, CsCl 20, EGTA 10, and HEPES 10 (pH 7.35 adjusted with CsOH). The HEK293 cells used in our experiments did not show significant persistent INa in the absence or presence of tetrodotoxin as previously described.11)12)
Results
Patient demographics
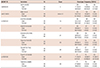
Table 2 shows patient demographics. The mean age is 56 years old, and the mean left ventricular ejection fraction was 65%. 9 patients were male. The mean sinus pause detected by surface ECG or Holter monitoring was 4.7 sec.
Gene sequencing
We sequenced all exons (exon 2-exon 28) of the SCN5A gene except for untranslated regions. Although we analyzed only 30 samples from Korean SSS patients, we identified 9 genetic variations, consisting of 2 synonymous, 6 non-synonymous, and 1 at the splicing donor site of the intron9-exon10 boundary (Table 3). Among these, 7 variations (G87A-A29A, IVS9-3C>A, A1673G-H558R, G3823A-D1275N, T5457C-D1819D, T5963G-L1988R, and C5129T-S1710L) have previously been reported in the American, Japanese, and Han Chinese populations, and their allele frequencies were similar to that in the Japanese or Chinese series. In addition, 2 novel variations (A3075T-E1025D and T4847A-F1616Y) were also found in two patients (Fig. 1). To compare with the control population, we analyzed normal samples using the same methods in order to determine whether the same variations existed in their genes. Five variations (G87A-A29A, IVS9-3C>A, A1673G-H558R, T5963G-L1988R, and T5457C-D1819D) were also found in the control group, however the novel genetic variations (A3075T-E1025D and T4847A-F1616Y) were not present in any of the 80 normal controls.
Three-dimensional model of sinlge nucleotide polyporphysm
We successfully generated a three-dimensional model of the SCN5A domain (amino acid residues 1525-1750), which revealed 8 alpha-helices and 7 loops or kinks. We were able to use this model to analyze one of the novel variations (F1616Y) found in this study, and one of the known variations (S1710L) (Fig. 2). The location of the S1710L variation was immediately after the 6th alpha helix. The long 6th loop started from this area. In addition, the serine to leucine change likely generated an extended 6th alpha-helical structure. F1616Y was located in the center of the second loop, which is well exposed. Three-dimensional modeling of the other variations was not successful due to a lack of available crystal structures for the homologous templates.
Patch-clamp recording
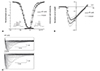
Figs. 3 and 4 show the representative tracings and time course of INa at a frequency of 20/min. The INa was induced by a repetitive depolarization pulse (to -10 mV for 300 ms) from a holding potential of -140 mV. All experiments were carried out at room temperature. The peak INa and voltage-dependency in cells transfected with mutant E1025D were not significantly changed compared with wild-type (Fig. 3). However, the voltage-dependency of the Na+ channel in mutant F1616Y-expressing HEK cells was significantly leftward-shifted compared with wild-type (Vh inactivation of F1616Y-delQ=-104.47 mV±0.21, n=7 vs. Vh inactivation of WT-delQ=-84.89 mV±0.09, n=12, p<0.005; Vh activation of F1616Y-delQ=-55.36 mv±0.22, n=8 vs. Vh activation of WT-delQ=-44.21 mV±0.17, n=7, p<0.005) (Fig. 4A). Furthermore, the peak INa in cells transfected with F1616Y demonstrated a 140% increase compared with wild-type (-335.13 pA/pF±24.04, n=8 vs -139.95 pA/pF±23.76, n=7, respectively, p<0.005) (Fig. 4B).
Discussion
In the present study, the genetic nucleotide sequence of SCN5A was determined in Korean SSS patients and compared with that of the controls. We found two novel genetic variations (A3075T-E1025D and T4847A-F1616Y). In addition, the normalized INa in mutant F1616Y-expressing HEK cells showed a leftward-shift in inactivation and activation, and the peak INa was increased.
The SCN5A gene consists of 28 exons and encodes a protein of 2016 amino acids, with a molecular mass of 227 kDa.13) In our patients with SSS, there were 9 sites of nucleotide change from exon 2 to exon 28 of the SCN5A gene. Among these, 2 sites (G87A-A29A and IVS9-3C>A) have been reported as genetic variations in a Western study,14) and T5457C-D1819D has already been reported as a variation or polymorphism without functional effects to the channel in Asian studies.15)16)17) Recently, Gui et al.18) reported that A1673G-H558R has variation-specific effects on SCN5A-related SSS. In addition, G3823A-D1275N, which is reported as a heterozygous loss-of-function variation, is related to reduced whole-cell currents due to impaired cell surface localization.19)
Since a synonymous nucleotide change in the SCN5A gene is unlikely to induce a functional change in the Na+ channel, a non-synonymous substitution was considered as a candidate for the variation associated with this disorder. The two new variations found in this study are non-synonymous and heterozygous changes. The variations may not affect the pathogenesis of SSS due to the opposite normal allele. Therefore, it is reasonable to suggest that the functional consequences of these changes should be assessed by measuring their effect on Na+ channel activity, allowing assessment of a direct relationship between the variations and disease.
We can deduce the functional effect of the variations by structural simulations, which provide direction for subsequent studies. In the present study, we obtained some information from a structural model of SCN5A on the functional effect of the variations S1710L and F1616Y. In the model, S1710L perturbs the Nav1.5 molecular structure from the 6th alpha helix - 6tth loop structural changes. It is also likely that the physiological consequences of this model would be misfolding of the Nav1.5 protein.20) However, F1616Y was located in the center of the second loop, which is well exposed. There is no significant steric hindrance or helix-packing problem with a change from phenylalanine to tyrosine at the 1616 site. Although there would be no significant structural changes with the F1616Y variation, one cannot exclude the possibility that it may be involved in protein-protein interactions, resulting in pathological consequences.
We used whole-cell configuration of the voltage-clamp technique to determine the effect of these genetic mutations. The mutation F1616Y led to a significant leftward-shift in the voltage-dependency of the Na+ channel. Karin et al.21) showed that some voltage-sensor Na+ channel mutations can lead to a 10 mV leftward shift of the steady-state fast inactivation curve, which enhances inactivation. In their study, the heterologous expression of the two Na+ channel mutations was shown to be important in pathogenesis by reducing the number of excitable channels. Similar to Lehmann-Horn's study,21) the mutation F1616Y makes a leftward-shift in the Na+ channel voltage-dependency, enhancing inactivation of the channel, and may be a cause of the pathogenesis of SSS, even in the heterologous mutation.
Most SCN5A mutations associated with sinus node dysfunction have shown a reduction in function or loss of function,4) but rare SCN5A mutations such as L212P have been reported to have gain of function.22) Mutations in SCN5A have been linked to LTQ3 due to an increase in persistent late INa (gain of function). However, the action potential in the sinoatrial node is largely calcium channel dependent.23) One possible explanation for how gain of function mutations in SCN5A could lead to sinoatrial node dysfunction is due to failure of the impulses to propagate into the adjacent atrial myocardium. The large hyperpolarizing shift in activation of the F1616Y channel reduces the threshold potential for activation, leading to increased INa. The hyperpolarizing shift of steady-state inactivation and delayed recovery from inactivation reduce Na+ channel availability and slow cardiac impulse conduction. Such opposing gating properties have been shown in the SCN5A mutation, G514C, responsible for isolated cardiac conduction disease.24) The G514C channel exhibits a depolarizing shift in both activation and inactivation. It is assumed that the balance of opposing gating abnormalities as demonstrated in G514C, under the influence of additional factors involved in impulse propagation, may elicit exit block in the sinoatrial node. In LQT3 families with 1795insD mutations, a persistent inward current or a negative shift in inactivation has been shown to impair the coupling of electrical events between the pacemaker cells and those surrounding them within the sinoatrial node. This led to impaired propagation of electrical activity from the sinoatrial node to atrial tissue.25)
In our study, the patients with novel genetic variations (A3075T-E1025D and T4847A-F1616Y) are all female and were diagnosed at a relative young age (<60 years), meaning that the genetic variation may be related to a clinical event. In particular, the patient with a genetic variation at T4847-F1616Y had a family history of SSS. Her mother had complained of SSS-related symptoms such as dizziness and palpitations in her forties. She was diagnosed by an electrophysiological study in her sixties. However, we could not confirm the presence of the genetic variation because she was already deceased. Therefore, we believe that this study on the functional consequences of the genetic variation is important.
A limitation of this study is the relatively small number of patients. In addition, we are not able to explain the exact relationship between the gain-of-function of F1616Y and SSS. However, Makita et al.22) reported gain of function of SCN5A in one family with congenital atrial standstill similar to our study. Finally, SCN5A may play a role in the SA node,4) therefore, we are planning to generate animal models for future research.
To our knowledge, this is the first study to determine a link between genetic variations in SCN5A and Korean patients with SSS. In this study, we demonstrated that there are 2 novel genetic variations (E1025D and F1616Y) in SCN5A in Korean patients with SSS.





 XML Download
XML Download