

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Melanoma is the most aggressive subset of skin cancer. Recent epidemiologic data from Europe and the United States indicates an alarming increase in incidence of melanoma in the last few decades.1 Analyzing three decades of data (1982–2011) from six populations with moderate-to-high melanoma incidence, Whiteman et al.2 reported that melanoma rates in the US, UK, Sweden, and Norway, have increased by more than 3 % annually. Prior to 2011, treatment of advanced melanoma was limited to dacarbazine (DTIC) and interleukin-2 (IL-2) with limited benefit achieved in a small subset of patients.3 Innovative melanoma treatments are now personalized based on the key oncogenes responsible for tumor development in combination with therapies stimulating the immune system to recognize aberrant cell lines.
Treatment response to molecular targeting in melanoma is dependent on each individual tumor's mutational status. According to the Cancer Genome Atlas network, the median mutation rate in melanoma is the highest of cancers thus far analyzed, about >10 mutations/Mb.4 As such, genetic sequencing for tumor mutational status is becoming increasingly applicable. Melanoma growth and progression can occur through constitutive activation of the mitogen-activated protein kinase (MAPK) and subsequent signaling through the RAS-RAF-MEK-ERK pathway.5 Mutations along melanoma's downstream signaling pathway, such as with RAS, BRAF, or MEK activating mutations, are often the culprits of this cancer's pathogenesis, and are thus also potential treatment targets.1 At diagnosis, patients with advanced cutaneous melanoma undergo tumor assays to assess the presence or absence of a BRAF mutation at codon V600 prior to treatment initiation. Although not commonly tested for, genetic sequencing for NRAS and KIT mutational status is also available.6
Melanoma progression and survival is dependent on a cancer cell line's ability to manipulate cell death pathways, evade immune surveillance, and survive in an unfavorable microenvironment.1 With the development of immunotherapy (e.g. anti CTLA4 antibodies and PD1 inhibitors), melanoma treatment is also targeting systems external to tumor cells that are thought to play a prominent role in intrinsic resistance. In the following review, we will explore melanoma inducing mutations, cell death pathways, and novel targeted therapy as we begin to understand mechanisms of resistance to treatment and survival. To understand the processes which induce cell death, the mutations that allow melanoma proliferation must first be explored.
TARGETING PATHWAYS OF PROLIFERATION IN MELANOMA
Gene mutations and mutation variants differ between melanoma subtypes as demonstrated by recent whole genome sequencing studies.78 The mechanism of mutation in cutaneous melanoma is propagated by UV radiation exposure, while acral and mucosal melanomas tend to have lower mutation burdens and different genes implicated in mutagenesis. For example, BRAF, NRAS, CDKN2A, and TP53 are mutations common in cutaneous melanoma while NRAS, BRAF, and NF1 mutations are more consistently found in acral melanoma. Additionally, KIT mutations are identified more often in mucosal and acral melanomas compared to cutaneous melanomas.7 Interestingly, Moon et al.8 found that the cytologic morphology differed between mutant subtypes: BRAF V600E mutants had round, epithelioid cells while NRAS and NF1 mutations presented with bizarre cells. The most common mutation associated with a UV signatures and high mutation rates was NF1, found in 17 percent of melanomas.7 Conversely, the most frequently identified mutation was BRAF V600 in acral melanomas.8 Considering the diversity of subtypes, whole genome sequencing becomes increasingly important in personalized treatment of melanoma.
1. c-KIT inhibition
KIT is a transmembrane receptor tyrosine kinase proto-oncogene expressed on melanocytes, as well as other sites within the body. When bound to its ligand, the downstream RAS signaling pathway is activated. When KIT is mutated, a loss of function results and the normal pathway of melanocyte development is interrupted, potentially leading to tumor development.3 Imatinib is a tyrosine kinase inhibitor which works by inducing apoptosis and inhibiting proliferation of tumor cells when an activating KIT mutation is present.9 In contrast to BRAF, mutations in KIT are widely distributed over the coding region, therefore functionality may vary. This also suggests some KIT mutations may be passenger mutations rather than true drivers of unchecked proliferation.3 The variability in response to treatment based on the nature of KIT mutation is further supported by the reported results of Guo et al.10 They found nine of the 10 patients who achieved a response to therapy had melanomas harboring a mutation in exons 11 or 13 of KIT, while only one of three patients with amplified KIT alone achieved a response to imatinib. Their results imply that sequencing with more selective molecular criteria at melanoma diagnosis may predict response to imatinib.
Clinical application of imatinib in gastrointestinal stromal tumors (GIST) is remarkably successful, as 95 % of these cancers exhibit KIT mutation. Over the course of time, it has been shown that resistance to Imatinib in GIST occurs when additional KIT mutations are acquired during treatment.11 Unfortunately, resistance mechanisms in melanoma are thought to be more complex and involve mutations in additional oncogenes such as NRAS in the MAPK and PI3K pathways. NRAS mutation in some melanoma patients was a poor predictor for response to Imatinib.9 Due to an overall low incidence of KIT mutation positive melanomas (29 percent of mucosal, 18 percent of acral, and 23 percent of sun damaged skin melanomas3), imatinib's clinical use is limited in melanoma.
2. BRAF inhibition
B-RAF is a proto-oncogene in the RAS/MAPK signaling pathway with >50% of melanomas harboring the mutation BRAF.1213 Mutations in BRAF gene interrupt normal cellular development and increase oncogenic potential through various mechanisms. In melanoma, the somatic V600E mutation results in a sustained transmission of cell growth signals that lead to uncontrolled cellular division. Multiple therapeutic inhibitors of B-RAF are FDA approved for melanoma treatment, including sorafenib, debrafenib, and vemurafinib. Sorafenib directly inhibits the V600E B-RAF mutation, by reversing activation of the downstream MEK pathway. This causes an anti-proliferative effect on tumor cells.14 Debrafenib, a potent ATP competitive inhibitor of BRAF kinase, exhibits activity against melanoma both with and without BRAFV600E mutations. It also has activity against melanoma brain metastases.14 Vemurafinib, on the other hand, prevents MEK phosphorylation exclusively in BRAF V600E mutant cells.
Recent data obtained in experimental melanoma cell models and human tumor samples exposed several mechanisms of resistance to BRAF including reactivation of the MAPK pathway with continued ERK activation and activation of the parallel signaling pathway PI3K-mTOR. Proposed salvage strategies for resistance included ERK inhibitors and pan-PI3K inhibitors.15 Furthermore, the tumor microenvironment seems to play a major role in response to molecular therapies. Preliminary evidence suggests that the oncogenic potential of BRAF (BRAFV600E) relates to immune escape and that blocking its action via the MAPK pathway can increase expression of melanocyte differentiation antigens (MDA). The goal of combination immunotherapy is to increase cytotoxic T cell recognition of these MDAs therefore enhancing antitumor response. Conversely, MAPK inhibition alone has a deleterious effect on immunogenic T cells.12 Given the prevalence of this mutation, BRAF inhibitors have a high potential for clinical application, however due to current rates of resistance their clinical effectiveness has been limited.14
3. Mek inhibition
MEK is a mitogen extracellular signal-regulated kinase, and MEK inhibitors work by acting downstream of the mutant BRAF and inhibiting cellular proliferations and tumor development. Trametinib, selumetinib and cobimetinub are several MEK inhibitors approved for treating BRAF positive metastatic melanoma.16 Trametinib is active against both MEK1 and MEK2,17 whereas cobimetinib is specific for MEK1.18 Like trametinib, selumetinib inhibits both MEK1 and MEK2. Similar to BRAF inhibitors, MEK inhibitors encounter problems with resistance, and combination treatments with two or more agents may enable improved treatment outcomes.19 The purpose of combination therapy is to delay the resistance that occurs with the reactivation of the MAPK pathway when BRAF or MEK inhibitors are used in monotherapy. The addition of a MEK inhibitor following the development of mutations is not effective, therefore upfront BRAF-MEK combination therapy is currently recommended. Published in 2015, the COMBI-d study further confirmed the success of combination therapy with a 29 % reduction in risk of death with median overall survival of 25.1 months for the combination arm versus 18.7 months in the dabrafenib only arm.20 Cobimetinib is also approved for the treatment of BRAF-mutated melanoma in combination with the BRAF inhibitor, vemurafenib.18
4. mTOR inhibition
mTOR, or mammalian target of rapamycin, is a kinase whose activation is strongly associated with melanoma, acting as a growth promoting factor involved in protein translation and cell growth. Through activation of mTOR, phosphorylation of p70 ribosomal S6 kinase occurs, which is the major target of rapamycin.21 When mTOR is phosphorylated upstream, it initiates a kinase cascade that eventually inhibits autophagy which is important for cell cycle regulation. mTOR is inhibited by two rapamycin analogues everolimus and temsirolimus, but clinical trials have been largely unsuccessful in the treatment of metastatic melanoma.22 Everolimus has not yielded good clinical success as a single agent, despite promising preclinical studies.23 Interestingly, everolimus in combination with Anti-PD-L1 treatment reduced tumor burden of renal cell carcinoma (RCC) in murine models by increasing tumor infiltrating lymphocytes and consequently the ratio of cytotoxic T cells.24 Further studies on combination therapy with PD1 checkpoint inhibitors may be beneficial in melanoma; see below discussion. Temsirolimus, which was historically used to treat RCC, appears to show promise in treating metastatic melanoma when used in combination with other treatments.25 However, temsirolimus induces autophagy. Autophagy promotes tumor survival so that temsirolimus' limits its own activity.26 We will discuss in the following section how the anti-melanoma drug chloroquine works by inhibiting autophagy.
TARGETING CELL DEATH PATHWAYS IN MELANOMA
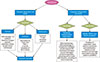
In the past few decades, evidence has accumulated on mechanisms of Regulated Cell Death (RCD). In Accidental Cell Death (ACD), cell demise occurs in the setting of physical stress such as high pressures, osmotic forces, and extreme temperatures. In contrast, RCD is programmed by predefined molecular machinery, and therefore can be modulated by targeted pharmacotherapy and genetic modification.27 Many of the mechanisms of cell death have overlapping signaling pathways which can be a challenge with treatment and emerging resistance. Various factors affect the fate of a cell to undergo apoptosis, autophagy, or necroptosis, see Fig. 1, including energy/ATP levels, the degree of damage or stress, and the presence of specific pathways inhibitors.28 Below we will discuss specific modalities of cell death as they apply to treatment of melanoma.
1. Apoptosis
Apoptosis is the earliest characterized process of cell death and the most well understood. It functions to maintain homeostasis of various cell processes, which means that the evasion of the apoptotic pathway is relevant to tumor formation and unchecked proliferation of melanoma cells. Apoptosis is mediated by both initiator and effector proenzyme, caspases, which propagate a proteolytic cascade ultimately terminating in cell death.2728 Apoptosis is initiated by three pathways, the Intrinsic, Extrinsic and Granzyme pathways, which converge at effector caspase 3 and ultimately result in DNA and protein breakdown. The intrinsic pathway, also known as the mitochondrial pathway, is initiated by unfavorable environmental conditions such as UV radiation, nutrient deprivation, oxidative stress, and replication stress. This process is regulated by proapoptotic and antiapoptotic molecules in the Bcl-2 family, which act to increase mitochondrial permeability and subsequent release of cytochrome c. On the other hand, the extrinsic pathway is death receptor mediated by FasL and TNFα binding to their respective receptors Fas and TNFR. Binding of these receptors activates an intracellular death domain that recruits proteins such as the TNF receptor-associated death domain (TRADD), the Fas-associated death domain (FADD), and the proenzyme caspase 8.29 The granzyme pathway is induced by cytotoxic T cells to allow the release of granzyme, a molecule that can cleave effector caspases.2728 Malignant melanoma cells can evade apoptosis through the following methods: disturbed balance of pro-apoptotic and anti-apoptotic proteins, reduced caspase function, and compromised death receptor signaling.29
Melanoma proliferation can also occur with dysregulation of several contributing modulators of apoptosis such as p53 protein and inhibitors of apoptosis proteins (IAPs). Avery-Kiejda et al.30 described how some target genes of p53 involved in apoptosis and cell cycle regulation are aberrantly expressed in melanoma cells and decreased expression of p53 correlated with proliferation of melanoma cells. IAPs are endogenous inhibitors of caspases and overexpression of these proteins has been associated with pharmacologic resistance in many cancers. Vucic et al.31 reported elevated expression of ML-IAP confers melanoma cell resistance to apoptotic stimuli and may contribute to the pathogenesis of tumor formation. The development of gene and immunotherapies to target BCL-2 family members, p53, IAPs, caspases, and other molecules involved in apoptosis is fundamental to the future treatment of melanoma.
2. Autophagy
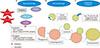
Autophagy is a cell death mechanism that involves self-degradation in response to nutrient stress. It is a survival mechanism, in that it selectively removes misfolded proteins and disposes of damaged organelles.32 The highly conserved process of autophagy involves engulfment of portions of cytoplasm and proteins by autophagosomes, which are then bound for lysosome fusion and degradation. The products of degradation then function as substrates for protein synthesis and energy production.33 Autophagy can be initiated by three different pathways: macroautophagy, microautophagy, and chaperone mediated autophagy.34 These mechanisms are further summarized in Fig. 2.
Autophagy is controlled by multiple pathways including the PI3K/Akt/mTOR pathway, often mutated in melanoma cell lines. When mTOR is phosphorylated upstream, it initiates a kinase cascade that eventually inhibits autophagy. With this mechanism, mTOR inhibitors have shown promise in modulating cell death in many solid tumors including melanoma. Conversely, the downstream effect of these inhibitors can lead to prosurvival of cancer cells by recycling damaged cell material and proteins for use by proliferating tumor cells.33 Xie et al.26 determined that melanoma cells are autophagy-dependent by demonstrating that knockdown of autophagy gene product ATG7 resulted in cell death. Recent studies have shown that inhibition of autophagy by chloroquine (CQ) has been employed to halt melanoma cell growth when used in combination with mTOR inhibitors. CQ inhibits autophagosome function leading to cytotoxicity in melanoma cells. It is unique in that this inhibition is independent of their BRAF mutational status. It was also found that combining CQ with echinomycin, a HIF-1α inhibitor, improved cytotoxicity in hypoxic conditions.35 In conclusion, malignant melanoma cells exhibit high levels of autophagy so therapeutic advances should aim at depriving tumor cells of this perpetuating energy source.
3. Necroptosis
Necroptosis, or regulated necrosis, is a process of cell death dependent upon RIPK1 and RIPK3 kinases which occurs independent of caspases. In contrast to apoptosis, necroptosis is often activated under conditions of insufficient caspase activation. The pathway by which necroptosis occurs is through the RIPK1/RIPK3/MLKL activation pathway. Caspases, which are essential to apoptosis execution, negatively regulate necroptosis through the cleavage of RIP1 and RIP3.28 When necroptosis is initiated, the plasma membrane permeability increases and damage associated molecular patterns (DAMPS) are released. Similar to apoptosis, necroptosis can be triggered by members of the TNF family through a different mechanism dependent on inhibition of caspase-8.36 The necroptosis pathway is often dysregulated in melanoma tumor cells due to lack of expression of RIPK3. Recent functional studies have also identified RIPK-1 signaling as a critical component in necroptosis induction. When RIP3K mutation was present, BRAF inhibitor Dabrafenib, but not Vemurafenib, inhibited necroptosis in melanoma cells. Given that RIPK3 expression can unmask necroptotic signaling, reactivation of this pathway may have therapeutic significance in metastatic melanoma.37
Often, low levels of death signals stimulate apoptosis while high levels of death signals induce necroptosis.28 Apoptosis is compromised in many types of solid tumors. As such, melanoma can acquire apoptosis-resistance to the typical anticancer agents. This resistance mechanism is problematic when using TNF-related apoptosis-inducing ligand (TRAIL), IAP inhibitors, and Bcl-2 inhibitors.38 Additional pharmacologic interventions to induce necroptosis as an alternate mode of programmed cell death could be beneficial in tumor treatment. One proposed challenge in selective initiation of necroptosis is how this would affect normal, non-malignant cells.28
4. Immunogenic cell death: Pd1 inhibitors, anti CTLA4 antibodies and CSF1r inhibitors
An important aspect of cellular immune defense against neoplasms such as melanomas involves the presentation and recognition of tumor antigens by a tumor specific T cell receptor. Both CTLA4 and PD1 receptors function as coinhibitory receptors on T-cells to inhibit tumor apoptosis and cytotoxic T cell action. Novel therapies target coinhibitory molecules which serve to dampen the immune response by downregulating the action of CD4 and CD8 cells. PD1 monoclonal antibodies, nivolumab and pembrolizumab, are approved for treatment of metastatic melanomas and act to strengthen the immune response to tumor antigens. A third immune modulator, imalimumab, was the first cytotoxic T lymphocyte antigen 4 (CTLA4) approved for combined use with vemurafenib for treatment of metastatic melanoma.39
Tumor associated macrophages (TAM) make up a considerable portion of tumor infiltrating immune cells in melanomas and play a complex role in the tumor microenvironment. Several studies have shown that macrophage abundance correlates with melanoma thickness, but correlation of density with cancer survival and mortality is unclear.40 Two main phenotypes of TAM exist, likely on a spectrum and have opposing roles in tumor progression. M1-like TAMs, or classically activated TAMs, have been identified as having antitumor functions and thus are targeted by anticancer therapies and gene targeted studies. M2-like TAMS, or alternatively activated TAMs, promote tumor growth by facilitating angiogenesis, cancer cell invasion, and metastasis. In recent years, research has focused on the immunosuppressive role of colony stimulating factor 1 (CSF1), its receptor CSF1r, and their relation to M2-like TAMS. One particular study showed CSF1 expression by melanoma cells may interfere with immune attack by IFNγ-secreting T cells when activated. The results of this study suggest that simultaneous inhibition of CSF1 and the CSF1R pathways could increase antitumor effectiveness of ‘CD8 T-cell function and immune checkpoint inhibitors’, which function less effectively in high intratumoral T-cell concentrations.40 In order to address the cancer associated immunosuppression in the microenvironment, combining PD1 inhibitors with CSF1R inhibitors may improve the clinical response of melanoma and decrease resistance to either treatment alone.
CONCLUSION
The use of mutation specific pharmacotherapy in melanoma has grown along with the understanding that resistance to targeted therapies can lead to the simultaneous emergence of resistant clones at many separate body sites, despite an initially positive therapeutic response. In this review, we discussed the complexity in pharmacological manipulation of melanoma with c-Kit, BRAF, MEK, and/or mTOR mutant cell lines. We also discussed melanoma evasion of cell death through modalities of RCD such as apoptosis, autophagy, and necroptosis. Lastly, we recognized the importance of immunomodulation though manipulation of the body's natural killing mechanisms, i.e. inducing cytotoxic T cell response to tumor cells and checkpoint inhibition. The melanoma mechanisms of cell death are summarized in Table 1.
Our hope is to identify resistance processes in order to improve the tumor response of current molecular and immunotherapy. As we begin to recognize tumor cell activation of alternate pathways, evasion of programmed cell death, and manipulation of the tumor microenvironment, it is increasingly important to grasp the complexity of personalized therapy in melanoma.

 XML Download
XML Download