

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Osteopetrosis is rare, heterogeneous, and inherited disorders of the skeleton characterized by increased bone mass and universal osteosclerosis. The disease was first described by the German radiologist Heinlich Albers-Schönberg in 1904 [1]. Osteopetrosis is related to congenital defect in the development or function of the osteoclast, thereby leading to generalized sclerosis of bone and reduction of bone marrow. Moreover, previous investigations reveal that osteoclast fail to produce the sufficient lysosomal enzymes for bone resorption in to extracellular space [2].
Several types are known with different models of inheritance and severity in osteopetrosis. Autosomal recessive osteopetrosis is known to be caused by loss-of-function mutation in genes including T cell immune regulator 1, ATPase H+ transporting V0 subunit a3 (TCIRG1), chloride voltage-gated channel 7 (CLCN7), osteoclastogenesis associated transmembrane protein 1 (OSTM1), pleckstrin homology and RUN domain containing M1 (PLEKHM1), sorting nexin 10 (SNX10), TNF superfamily member 11 (TNFSF11), TNF receptor superfamily member 11a (TNFRSF11A), etc. Intermediate recessive osteopetrosis induced by type II carbonic anhydrase deficiency is usually found during childhood and the severity of the disease is variable [3]. The most aggressive forms tend to have autosomal recessive inheritance, whereas the benign types are observed in adults and inherited in an autosomal dominant manner [3]. Comparing to different types of osteopetrosis with life-threatening complications in the first decade of life, type II autosomal dominant osteopetrosis (ADO II also known as Albers-Schönberg disease) is more benign condition principally affecting adolescents and adults. Life span in the adult onset form is also usually normal [2]. However, it causes commonly diffuse osteosclerosis predominating in spine and pelvis, thus promoting to development of the classic sandwich vertebra appearance in patients with ADO II [4].
ADO II is the relatively common form of osteopetrosis and has an incidence of 1 in 20,000 births. To date, mutations in at least 10 genes have been identified to induce failure of osteoclast differentiation or function in humans [2]. Above 70% of patients harbors heterozygous dominant negative mutations of the CLCN7 gene [5]. Mutations in CLCN7 gene lead to dysfunction of chloride channel in osteoclast, resulting in ADO II. CLCN7 plays a key role in lysosomal acidification which is crucial in dissolving bone mineral and organic bone matrix by osteoclast [6].
Herein, we report a case of a 68-year-old woman with ADO II who presented with pain in hip joint. We also detect a heterozygous mutation in CLCN7, the first report in Korea, through whole exome sequencing using genomic DNA of the patient. The mutation of the patient was also regarded to have a pathogenic effect, which was predicted by in silico analysis of the effect of amino acid substitution.
METHODS
Ethics statement
This study was reviewed and approved by the Institutional Review Board of Chungnam National University Hospital (CNUH 2015-09-042). Written informed consent was obtained from the subject before participating in the study. Current research was also conducted in accordance with the precepts of the Declaration of Helsinki.
DNA preparation
A 10 mL of peripheral blood sample was obtained from the patients. Genomic DNA was extracted in the whole blood samples of the patient with QiaAmp DNA blood mini kit (Qiagene, Valencia, CA, USA).
Whole exome sequencing
After informed consent was acquired, whole exome sequencing was conducted using genomic DNA of the patient through the service from Macrogen (Daejeon, Korea). The human exome capture by the SureSelect V6-Post kit (Agilent, Santa Clara, CA, USA) was used. Paired-end sequencing was performed using Illumina Hiseq 2500 platform (Illumina, San Diego, CA, USA). Data analysis was conducted by the Macrogen exome sequencing pipeline. Briefly, the raw data were mapped to the reference genome (hg19) with the BWA v0.7.12, and variants were called using GATK v3.4.0. Variants were annotated with ANNOVAR. The population frequencies of single nucleotide variants (SNVs) and small insertions and deletions (indels) were annotated with dbSNP, ESP6500, and 1000 genome database. The mean depth of the target regions was 83.1 reads. The 96.7% of target sequences were covered with >20 independent reads. The throughput of the exome sequence is available upon request.
Polymerase chain reaction and Sanger sequencing
Genomic DNA was subjected for polymerase chain reaction (PCR) and Sanger sequencing on CLCN7 exon 4 using following primers: forward, 5′-TTG CCT TTT TCT GCC ACA G-3′; reverse, 5′-GAG GAG GAG GGA GGA GGT G-3′. The PCR reaction was performed using the following protocols: denaturation was conducted for 1 minute at 94℃, followed by 35 cycles of 94℃ for 30 seconds, 61℃ for 30 seconds, and 72℃ for 1 minute, with a final 7-minute extension at 72℃. The amplified 400-bp PCR products were purified using ExoSAP reagent, and forward and reverse sequencing were performed using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems Inc., Foster City, CA, USA) and an ABI 3730 XL automatic sequencer (Applied Biosystems) using the same primers.
Prediction of functional effects of human single nucleotide polymorphism
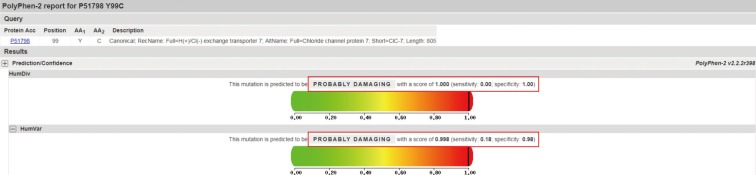
Polymorphism Phenotyping v2 (PolyPhen-2) as a tool for annotating coding nonsynonymous single nucleotide polymorphisms was used to predict the functional effects of amino acid substitution on the structure and function of a human protein [7]. PolyPhen-2 helps us to hypothesize the clinical significance of the mutations in humans. This program evaluates the mutations as benign, possibly damaging, or probably damaging based on pairs of false positive rate thresholds (http://genetics.bwh.harvard.edu/pph2/index.shtml).
RESULTS
Clinical report
A 68-year-old female patient visited department of orthopedics due to pain in hip joint. She complained of continuous both hip pain for 3 years prior to hospital visit and was unable to walk independently 3 months ago. She underwent total knee arthroplasty due to osteoarthritis 2 years ago, and then has been in a wheelchair since 3 months ago. She was scheduled to have a total hip replacement operation. There was no family history of known symptoms and diseases of the skeletal system. Her height was 150 cm, and body weight was 45 kg. Physical examination revealed that blood pressure 120/70 mm Hg, pulse 70 beats per minute, respiratory rate 18 breaths per minute, body temperature 36.8℃. Intraoral examination revealed no gingival inflammation or recession and abnormalities in teeth of the patient.
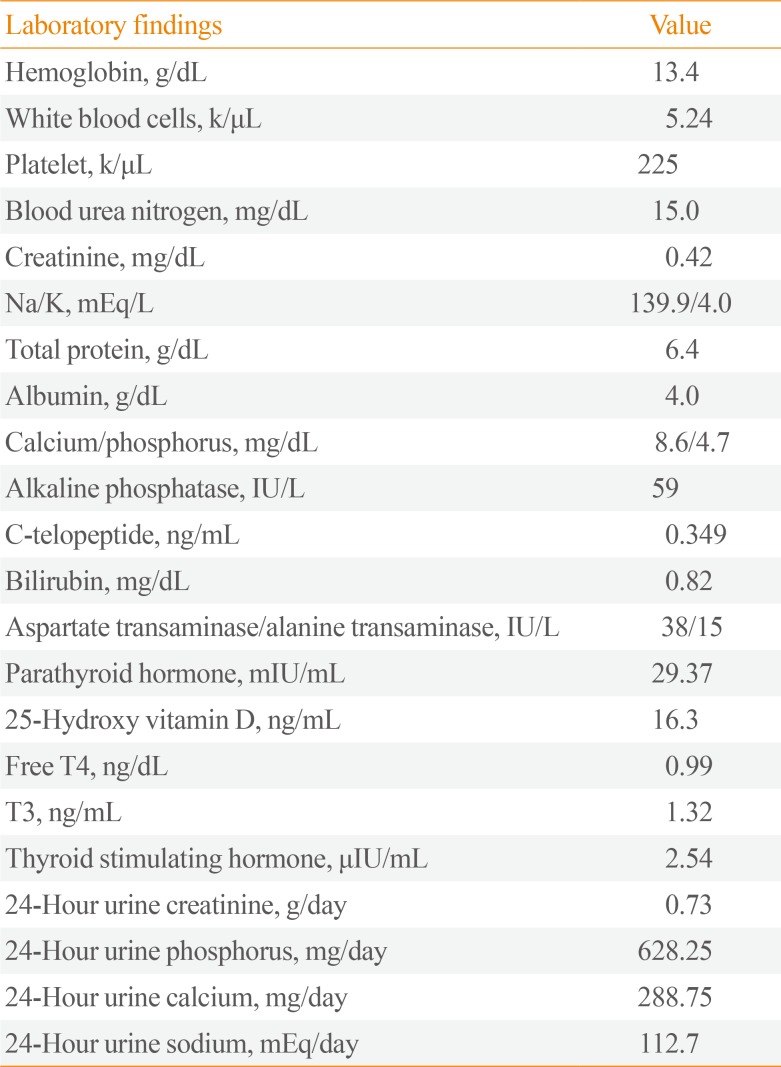
Laboratory findings of the patient showed hemoglobin of 13.4 g/dL, white blood cells 5.24 k/µL, and platelet count 225 k/µL. Renal function and serum chemistry were within normal limit: blood urea nitrogen 15.0 mg/dL, creatinine 0.42 mg/dL, Na/K 139.9/4.0 mEq/L, total protein 6.4 g/dL, albumin 4.0 g/dL, calcium/phosphorus 8.6/4.7 mg/dL, alkaline phosphatase 59 IU/L, and C-telopeptide 0.349 ng/mL. Liver function test were normal: serum bilirubin 0.82 mg/dL, and aspartate transaminase/alanine transaminase 38/15 IU/L. Serum parathyroid hormone was 29.37 mIU/mL (reference range, 10 to 65) and 25-hydroxy vitamin D was 16.3 ng/mL. Thyroid function test showed normal: free thyroxine 0.99 ng/dL, triiodothyronine 1.32 ng/mL, and thyroid stimulating hormone 2.54 µIU/mL. Serum protein electrophoresis was normal and immunofixation electrophoresis also revealed no evidence of monoclonality in serum protein. Twenty-four-hour urinalysis results were all within normal limits: creatinine 0.73 g/day, phosphorus 628.25 mg/day, calcium 288.75 mg/day, and sodium 112.7 mEq/day. All laboratory findings were summarized in Table 1.
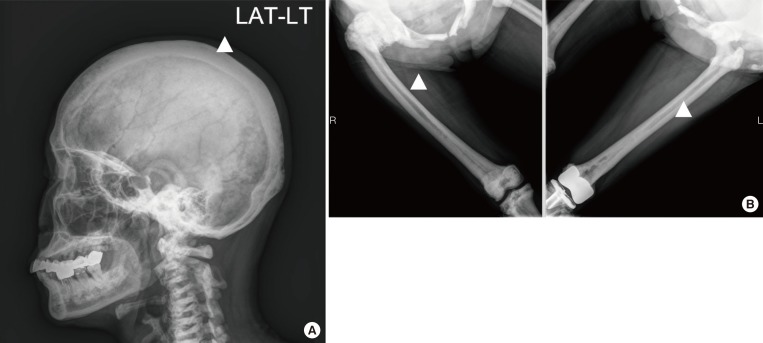
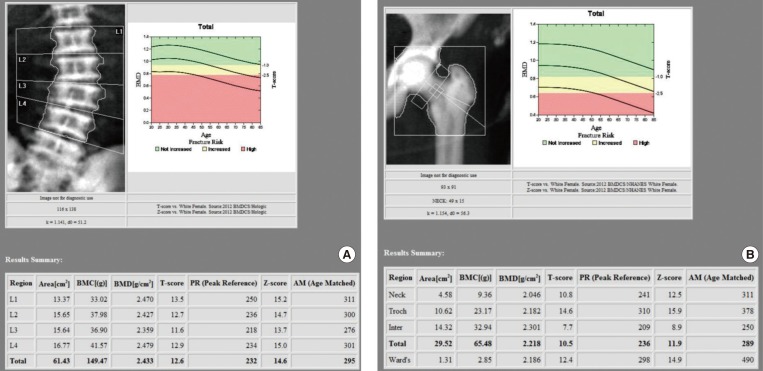
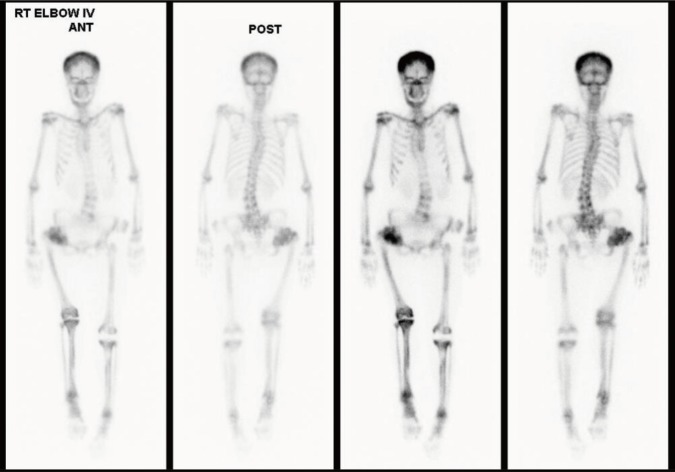
Simple X-ray for skull, spine and pelvis, bone scan, and dual-energy X-ray absorptiometry were included in radiologic investigations. Lateral skull view on radiograph revealed thickening of the inner and outer cortical tables (Fig. 1A). The base of the skull appeared relatively higher radiodensity with loss of trabecular pattern (Fig. 1A). Lateral view of lumbar spine also showed sandwich vertebra appearance as well as increased radiodensity. Moreover, simple radiography of pelvis shows generalized increased bone density and “bone-in-bone” appearance in her right femur head (Fig. 1B). The bone mineral density of the patient was shown to be remarkably increased using dual-energy X-ray absorptiometry (Fig. 2). Whole-body bone scintigraphy revealed abnormal uptake in proximal epiphysis of both humeri, tibias, and fibulas. High uptake was also found in tight femoral head and acetabulum, but kidneys and bladder were not visible, suggesting bone superscan (Fig. 3).
Genetic analysis with whole exome sequencing and direct DNA sequencing
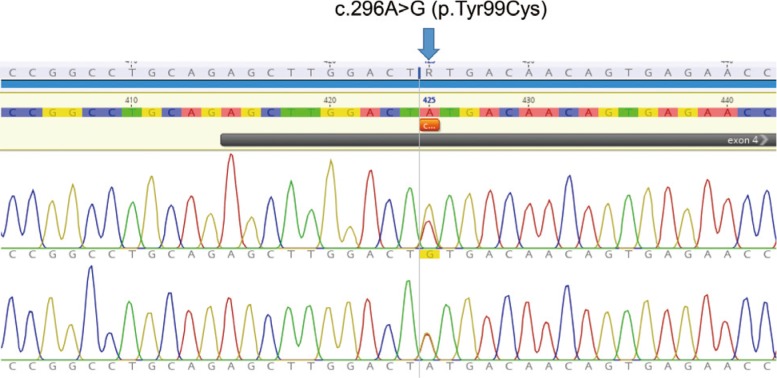
We obtained 98,367 variants from the patient using whole exome sequencing. Among them, the number of synonymous variant, missense variant, and frameshift variant was 12,074, 11,518, 312, respectively. Three variants were identified in coding regions of CLCN7 gene, including two synonymous variants and a heterozygous missense c.296A>G (Tyr99Cys, rs387907576) SNV at exon 4 of CLCN7. This variant has been reported in ClinVar database as pathogenic/likely pathogenic, and has not been reported in a population databases including 1000 Genome and ESP6500. All genetic data regarding the variants are available upon request. Additionally, direct sequencing of the CLCN7 gene showed a heterozygous mutation at the same position (Fig. 4). This tyrosine residue is also highly conserved across species, for example, human, rhesus, mouse, chicken, zebrafish, frog, and lamprey (Fig. 5). Moreover, PolyPhen-2 prediction result of probable damaging was reported based on in silico analysis of the effect of amino acid substitution (Fig. 6). The in silico prediction by SIFT was damaging (score, 0.000) and MutationTaster predicted it as disease causing variant.
DISCUSSION
The diagnosis of ADO II was usually based on genetic analysis, typical clinical, hematological parameters, and radiographs of the spine and pelvis. ADO II is the relatively common type of osteopetrosis with an incidence of 1 in 20,000 births [2]. Although our patient was a 68-year-old female, the incidence of ADO II is similar in men and women [5]. ADO II shows highly disease course heterogeneity ranging from asymptomatic early stage patients to severely debilitating phenotypes. The major clinical manifestations of ADO II are long bone fracture, osteoarthritis, osteomyelitis, dental abnormalities, cranial nerve compression, and moderate hematologic failure [8]. However, in the current study, the patient has only hip pain and osteoarthritis without history of other skeletal manifestations. Bone marrow dysfunction is relatively uncommon clinicopathologic finding in ADO II [9], and complete blood counts of our patient was also within normal limits.
For induction of bone resorption, the osteoclast should be differentiated from its precursors, form the ruffled borders with attachment to the bone surface, and remove degraded the organic bone matrix and calcium through secreting acid and proteases [10]. CLCN7 provides the chloride transport required for an efficient proton pumping by the H+-ATPase of the osteoclast ruffled membrane [11]. Therefore, mutations in the chloride channel 7 gene lead to acidification failure of extracellular lysosome between osteoclast and bone, resulting in a defect in bone resorption [12]. Subsequently, these studies on patients with ADO II have provided the potential value of the development of CLCN7 inhibitor for treatment of osteoporosis and metabolic bone diseases. Treatment with CLCN7 inhibitors prevents bone resorption without changes in bone formation in ovariectomized rat model [13], but further studies using CLCN7 inhibitors have not been performed. However, cathepsin K, acid-activated protease mediating type I collagen cleavage in the resorption lacunae, has been an attractive target for the drug development of osteoporosis. Odanacatib as a cathepsin K inhibitor was recently developed and showed reduction in fractures in postmenopausal women, but significantly higher risk for stroke in the odanacatib group was detected in a large phase III clinical trial, leading to early termination of clinical trial [14].
CLCN7 has 12 transmembrane hydrophobic domains and two cytosolic domains, which are comprised of 25 exons spanning over 30 kb in the human genome [1215]. CLCN7 forms a homodimer having two homologous subunits, consisting of eighteen intramembrane α helices, four chloride ion binding sites, and two cystathionine beta synthase domains [16]. In ADO II, pathogenic variants of CLCN7 comprise nonsense variants, frameshifts, insertions, deletions, and missense variants. Approximately 70% of patients with ADO II harbors autosomal dominant mutations of the CLCN7 gene [3]. The mutation c.296A>G of our patient is located in exon 4 of the intramembrane α helices in CLCN7. We found that the result of probable damaging was reported based on in silico analysis of the effect of amino acid change (Fig. 6). Because this variant was reported as pathogenic/likely pathogenic in ClinVar database, absent in population databases, and multiple lines of computational evidences support a deleterious effect, we regarded this variant as a pathogenic variant [17]. Previous investigations demonstrated the variation in age at diagnosis of ADO II. In a Chinese osteopetrosis family, ages of patients with ADO II ranged from 3 to 29 years [1819]. In contrast, the age at diagnosis of ADO II was 68 years in our patient. Moreover, Chinese and Korean patients with mutation c.296A>G in CLCN7 showed a substantial difference in bone phenotype. This suggests that no straightforward genotype-phenotype correlation could be established between CLCN7 variants and disease characteristics.
In conclusion, we report on a germline CLCN7 mutation in adult female patient with ADO II. This is the first description genetically confirmed with the heterozygous missense c.296A>G (Tyr99Cys) mutation in a Korean patient with ADO II.

 XML Download
XML Download