

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Dear Editor,
X-linked retinoschisis (XLRS; MIM 312700) is one of the most common genetic causes of progressive retinal degeneration in young males [1]. The main clinical feature of XLRS is schisis (splitting) of the retinal layers, leading to visual failure early in life [1]. XLRS is caused by mutations in RS1, located on chromosome Xp22.13 and encoding the RS1 protein, which plays an important role in the cellular organization of the retina [2]. Of the Korean cases of XLRS based on molecular studies [345], the most frequent RS1 variant was the missense type (88%, 14/16) [4]. We report a case of late-onset XLRS in a Korean patient harboring a novel pathogenic RS1 frameshift variant (c.362delA) and compare his clinical and molecular characteristics with those of patients from previous studies. To our knowledge, this is the first genetically confirmed case of late onset XLRS with a pathogenic frameshift variant in the Korean population. Institutional Review Board approval was exempted for this study.
A 41-year-old man (II-1; Fig. 1A) visited Soonchunhyang University Bucheon Hospital in September 2016 for check-up of deteriorating visual acuity over the past four years. His best-corrected visual acuity was 0.5 in both eyes. Ophthalmoscopic examination revealed bilateral wheel-like maculopathy (Fig. 1B). Spectral domain optical coherence tomography showed pronounced inner retinal cysts in the foveal area (Fig. 1C). Electroretinogram (ERG) documented marked reduction of the b wave (Fig. 1D). The patient's brother (II-2, Fig. 1A) also had poor visual acuity, and he had been diagnosed as having cystic maculopathy. Both brothers received an intravitreal corticosteroid injection to reduce macular edema. Neither had any vision-threatening complications of XLRS, including vitreous hemorrhage or retinal detachment, up to May 2018, the last follow-up visit. Neither of their parents had maculopathy.
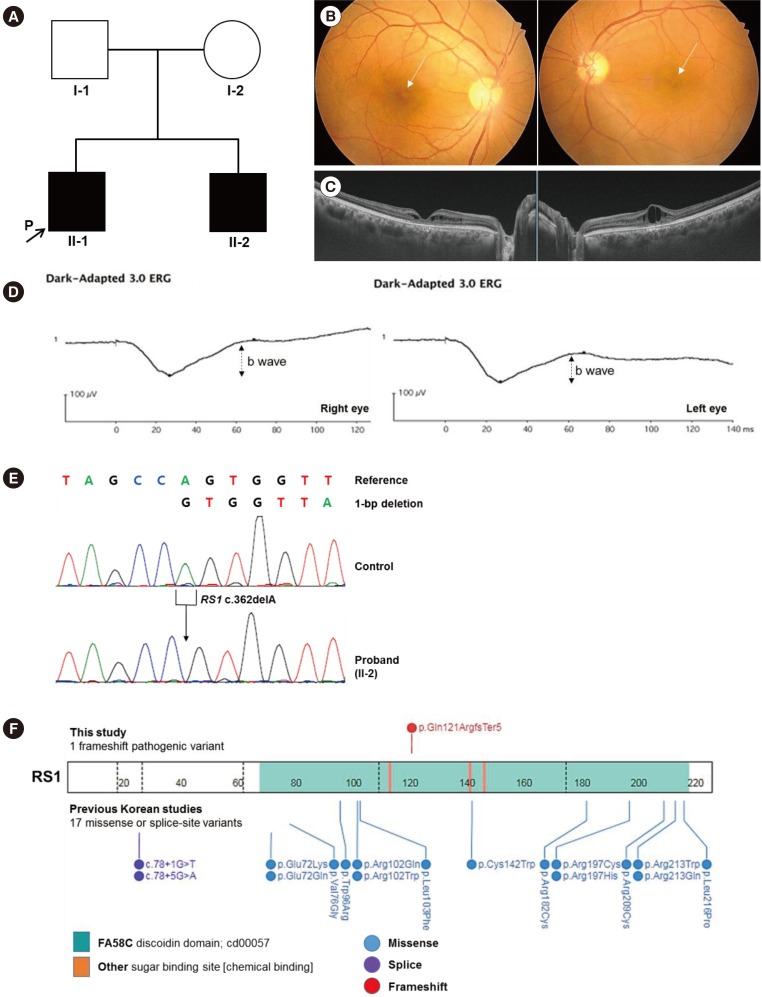 | Fig. 1A family affected by a novel pathogenic variant in RS1. (A) Pedigree of the family showing two related cases of XLRS. The arrow indicates the patient. A black symbol indicates clinically affected family members. (B) Fundus photograph showing typical wheel-like maculopathy (arrows). (C) Spectral domain optical coherence tomography showing marked retinoschisis in different retinal layers. (D) ERG showing reduced amplitude of b waves in both eyes, which is a key feature of XLRS. (E) A hemizygous pathogenic variant of c.362delA (p.Gln121ArgfsTer5) in RS1 was identified by Sanger sequencing. (F) Mutational landscape of RS1 in Koreans. The site of the pathogenic frameshift variant we described is indicated above the bar. Sites of previously reported pathogenic variants observed in Korean studies are indicated below the bar [345].Abbreviations: XLRS, X-linked retinoschisis; ERG, electroretinogram.
|
After obtaining informed consent, we extracted genomic DNA from the patient's peripheral blood leukocytes. For RS1 analyses, PCR (using primers: exon 1F, 5′-GGTTAACTTGATGGGGCTCA-3′; exon 1R, 5′-AACTGGAAAGCCATCCACAC-3′; exon 2F, 5′-AGCATCTGCGGATGTTTTTC-3′; exon 2R, 5′-TGTTGGGATTACAGGCATGA-3′; exon 3F, 5′-TCAATTTGGCCATTGTAGCA-3′; exon 3R, 5′-GGAGAAAACCCGCATTAACA-3′; exon 4F, 5′-GCAAAGCAGATGGGTTTGTT-3′; exon 4R, 5′-TTCCCAGGTTCAAGCAATTC-3′; exon 5F, 5′-GGAGACAAGGCTCAGACTGC-3′; exon 5R, 5′-ACAGAGGGCAGTGACAGGAG-3′; exon 6F, 5′-GTTCCAGATGTCCCAAGCAT-3′; exon 6R, 5′-TGCGAAATATAGCCCTGTCC-3′) was followed by sequencing. Sequences were compared with the reference sequence for RS1 (NM_000330.3). A novel hemizygous 1-bp deletion (c.362delA) in RS1, which was predicted to result in a frameshift and premature termination of the RS1 protein (p.Gln121ArgfsTer5), was identified (Fig. 1E). The c.362delA variant is located in the discoidin (DS) domain of the RS1 protein, a crucial region for its functional activity (Fig. 1F) [1]. This variant has not been previously reported and is not found in the Korean Reference Genome Database [6] or the >138,000 exome and genome sequencing data deposited with the Genome Aggregation Database (gnomAD) [7]. According to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines [8], this variant is classified as pathogenic based on one very strong piece of evidence (PVS1) and two moderate pieces of evidence (PM1, PM2). Family study could not be conducted.
To date, a total of 223 different disease-causing variants of RS1 responsible for XLRS have been reported worldwide in the Human Gene Mutation Database [9]. The majority are missense (52%, 116/223) with significant clustering within the DS domain (87%, 101/116); however, nonsense (11%, 25/223), small deletions (11%, 25/223), splice-site (10%, 22/223), and insertions (4%, 10/223) have also been observed with random distribution throughout the protein [9]. The missense type was also predominant in previously reported Korean patients [4] with varying clinical severity, and all variants were located within the DS domain of the RS1 protein (Fig. 1F). To examine the genotype-phenotype correlation in patients with XLRS, we compared the clinical and molecular features of our patient with those of previous reports (Table 1). Vincent et al. [10] suggested that pathogenic variants that putatively cause protein truncation result in greater clinical severity and an abnormal ERG. However, our patient with an RS1 frameshift variant showed typical foveal schisis and an electronegative ERG, but no peripheral schisis or ocular comorbidities. In addition, he was diagnosed as having XLRS at a late age compared with other patients, who were generally diagnosed with XLRS in the first decade of life. Additional clinical and molecular studies involving patients with XLRS are needed to determine the relationship between genotype and phenotype expression and to elucidate the pathophysiology of XLRS.
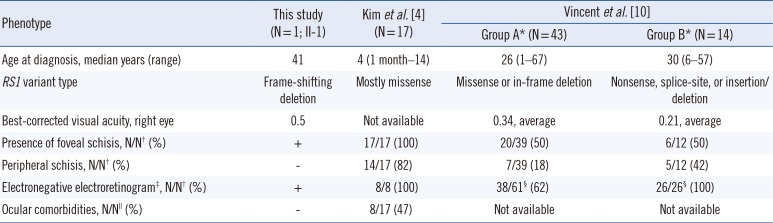
Table 1
Clinical characteristics of the present patient and previously reported patients with X-linked retinoschisis
| Phenotype | This study (N=1; II-1) | Kim et al. [4] (N=17) | Vincent et al. [10] | |
|---|---|---|---|---|
| Group A* (N=43) | Group B* (N=14) | |||
| Age at diagnosis, median years (range) | 41 | 4 (1 month–14) | 26 (1–67) | 30 (6–57) |
| RS1 variant type | Frame-shifting deletion | Mostly missense | Missense or in-frame deletion | Nonsense, splice-site, or insertion/deletion |
| Best-corrected visual acuity, right eye | 0.5 | Not available | 0.34, average | 0.21, average |
| Presence of foveal schisis, N/N† (%) | + | 17/17 (100) | 20/39 (50) | 6/12 (50) |
| Peripheral schisis, N/N† (%) | − | 14/17 (82) | 7/39 (18) | 5/12 (42) |
| Electronegative electroretinogram‡, N/N† (%) | + | 8/8 (100) | 38/61§ (62) | 26/26§ (100) |
| Ocular comorbidities, N/N∥ (%) | − | 8/17 (47) | Not available | Not available |
*Group A comprised 43 patients with missense or in-frame deletions in RS1; Group B comprised 14 patients with nonsense, splice-site, or frameshifting insertions/deletions in RS1; †N/N indicates the number of positive patients out of the total number of examined patients; ‡An electronegative electroretinogram has classically been defined as an electroretinogram in which the a-wave amplitude is normal, but the b-wave amplitude is reduced; §N/N indicates the number of positive eyes out of the total number of examined eyes; ∥Vitreous hemorrhage or retinal detachment.
![]()
Clinical diagnosis of XLRS can be challenging because of the variable phenotypic presentation and limited correlation between variant type and disease severity or progression. This report will contribute to a better understanding of the genetic background in Korean patients with XLRS.
 XML Download
XML Download