

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Chlorpromazine (CPZ) has long been used in clinical practice as an antipsychotic drug [2631]. The therapeutic effects of CPZ act via inhibition of calcium-(Ca2+)-dependent phospholipase A2 and are believed to occur via binding to calmodulin (Ca2+-binding messenger protein) [14]. Excessive stimulation of Ca2+-activated processes, such as those catalyzed by nonlysosomal proteases, endonucleases and phospholipases, may be the predominant cytotoxic event for a variety of toxic insults [28].
Oxidative stress and products of lipid peroxidation (LPO) are implicated in the pathophysiology of various neurological disorders. Chronic treatment with neuroleptics increases free radical production and oxidative stress [5]. Generation of reactive oxygen species (ROS) and reactive nitrogen species is known to damage various cellular components and thereby contribute to cellular dysfunction [33]. The products of LPO react with residues on amino acids such as cysteine or lysine and disturb protein function. It is well known that antipsychotics increase oxidative stress by altering the activity of antioxidant enzymes and causing oxidative damage, particularly LPO in the brain [30]. Antioxidant defense (AOS) mechanisms play the primary role in tissue repair and include enzymes that scavenge ROS (superoxide dismutase [SOD], catalase [CAT], glutathione peroxidase [GPx], glutathione reductase [GR]), but also nonenzymatic biomolecules (glutathione [GSH]).
In addition to a number of important physiological properties related to central nerve system (CNS) homeostasis, astrocytes are involved in regulation of the brain microenvironment. Activation of microglial and astroglial cells is a sensitive marker of the pathological processes occuring in the cellular microenvironment of the CNS [24].
Since CPZ induces oxidative stress by activating various reactive species that lead to tissue damage, our the study pointed to the potentially protective effect of (4-aminobutyl) guanidine, agmatine (AGM) and its ability to correct changes in indicators of oxidative/nitrosative stress induced by CPZ. AGM is the decarboxylation product of arginine and an intermediate in polyamine biosynthesis [35]. It is well known that AGM is a putative neurotransmitter synthesized in the brain, stored in synaptic vesicles, accumulated by uptake, released by membrane depolarization, and inactivated by agmatinase [32].
Based on this background, this study was conducted to investigate the role of ROS, efficiency of antioxidant protection, and importance of glial activation in the rat forebrain cortex 48 h after acute CPZ and AGM administration.
Materials and Methods
Animals
Experimental animals were treated according to the Guidelines for Animal Study, No. 282-12/2002 (Ethics Committee of the Military Medical Academy, Belgrade, Serbia and Montenegro). The experiments were performed on adult male Wistar rats weighing approximately 230 g that had been randomly divided into four groups. All animals were housed in cages under standardized conditions (ambient temperature of 23 ± 2℃ with 55 ± 10% humidity and a light/dark cycle of 13/11 h). The rats were given a commercial rat diet and tap water ad libitum.
Experimental procedure
The experiment consisted of four experimental groups of 10 rats each that received the following: normal saline intraperitoneally (i.p.) as a control (CG group); a single daily dose (38.7 mg/kg body weight [BW] i.p.) of CPZ-HCl (Medisca, Italy) (CPZ group); 38.7 mg/kg BW CPZ-HCl i.p. followed by 75 mg/kg BW i.p. daily doses of AGM (CPZ+AGM group); and a single daily dose (75 mg/kg BW) of AGM i.p. (AGM group). Animals were sacrificed by decapitation at 48 h after the appropriate treatment, after which the brain was excised and stored at −20℃.
Determination of CPZ concentration
The concentration of CPZ was determined by high performance liquid chromatography-tandem mass spectrometry (HPLC MS/MS) [19]. Briefly, 4 mL of acidic acetonitrile was added to 1 g of brain tissue and homogenized using an T18 digital ULTRA-TURRAX (IKA-Werke, Germany) homogenizer, then centrifuged for 10 min at 1,372 × g. Subsequently 6 mL of a 10% NaCl solution added to the supernatant. The samples were then purified on a C-18 column that had been preconditioned with 5 mL of methanol, folowed by 5 mL of water. After the sample extract was loaded onto conditioned SPE colums and passed trough, the columns were washed with 1 mL 0.01 M H2SO4. CPZ was then eluted from the columns with a 2 × 3 mL mixture of acidic acetonitrile and methanol (50:50) eluate and evaporated under a nitrogen stream, after which the residue was dissolved in 1 mL of acidic acetonitrile and methanol (50:50). The method was performed on an HPLC MS/MS Waters Acquity with TQD detector using a stock standard solution of CPZ prepared in methanol (concentration 0.897 mg/mL) and standard working solutions prepared by diluting the stock standard solution in the mobile phase.
Biochemical analyses
The forebrain cortex was dissected on ice, after which each tissue slice (approximately 0.1 g) was transferred separately into cold, buffered sucrose (0.25 M/L sucrose, 0.1 mM/L ethylenediaminetetraacetic acid [EDTA] in sodium-potassium phosphate buffer, pH 7.2). Aliquots (1 mL) were placed into a glass tube homogenizer (Tehnica Zelezniki Manufacturing, Slovenia) with a Teflon pestle at 1,000 × g for 15 min at 4℃. The supernatant was then centrifuged at 2,500 × g for 30 min at 4℃. The resulting precipitate was suspended in 1.5 mL of deionized water. Subcellular membranes were solubilized in hypotonic solution by constant mixing for 1h using a Pasteur pipette. Thereafter, homogenates were centrifuged at 2,000 × g for 15 min at 4℃ and the resulting supernatant was used for analysis [17]. The total protein concentration was estimated with bovine serum albumin as a standard [23].
Lipid peroxidation in the forebrain cortex was measured as tiobarbituric acid reactive substances production (TBARS) using the method described by Girotti et al. [15]. Data were expressed as nmol/mg proteins. Superoxide anion (O2•−) content was determined by the reduction of nitrobluetetrazolium in alkaline nitrogen saturated medium. Kinetic analysis was performed at 550 nm [4]. The results were expressed as nmol red nitro blue tetrazolium/min/mg proteins.
Total GSH (GSH + 1/2 GSSG, in GSH equivalents) content was determined using a DTNB-GSSG reductase recycling assay. The rate of formation of 5-thio-2-nitrobenzoic acid (TNB), which is proportional to the total GSH concentration, was followed spectrophotometrically at 412 nm [2]. The results were expressed as nmol/mg proteins.
SOD (EC 1.15.1.1.) activity was measured spectrophotometrically as the inhibition of epinephrine spontaneous auto-oxidation at 480 nm. The kinetics of sample enzyme activity was followed in a carbonate buffer (50 mM, pH 10.2) containing 0.1 mM EDTA after the addition of 10 mM epinephrine [39].
Catalase activity was determined by the spectrophotometric method. Ammonium molybdate forms a yellow complex with H2O2 and is suitable for measuring both serum and CAT activity in the tissue [16]. Kinetic analysis was performed at 405 nm. CAT activity was defined as µmol H2O2 reduced per minute (µmol H2O2/min). Data were expressed as U/mg proteins.
A ransel kit for the determination of GPx (Randox Laboratories, USA) was used to indirectly measure the activity of GPx [40]. The oxidation of NADPH to NADP+ is associated with a decrease in absorbance at 340 nm. Enzyme activity of GPx was defined as µmol NADPH oxidized per minute (µmol NADPH/min) and the results were expressed as U/mg proteins. The method for determining the activity of the GR is based on the ability of GR to catalyze the reduction of GSSG to GSH by oxidation of the coenzyme NADPH to NADP+ [13]. In the reaction, we used 100 mmol NAD+ as a standard. Enzyme activity was defined as µmol NADPH oxidized per minute (µmol NADPH). The results were expressed as U/mg proteins.
Reagents
All chemicals used in this study were of analytical grade. DTNB, NaH2PO4, ammonium molybdate, ammonium acetate, NADPH and NADH were purchased from Merck (Germany). Na2HPO4 × 2H2O, TCA, nitroblue-tetrazolium, gelatin, Na2CO3, NaHCO3, epinephrine, EDTA, NAD+, methanol and GSSG (oxidized form) were purchased from Serva-Feinbiochemica (USA). TBA was purchased from ICN Biomedicals (Ohio) and acetonitrile was purchased from Backer J.T., Deventer (the Netherlands). GSH reductase (EC 1.6.4.2; Type III, from yeast [9001-48-3]) and analytical standard for CPZ (catalog No. C8138) were purchased from Sigma-Aldrich (USA). Saline solution (0.9% w/v) was purchased by the Hospital Pharmacy (Military Medical Academy, Belgrade, Serbia). All drugs solutions were prepared on the day of the experiment.
Immunohistochemistry
For immunohistochemical analyses, brains (four per group) were quickly removed and fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (PBS) at pH 7.4 for 12 h at 4℃. For cryoprotection, brain tissue was transferred into the graded sucrose (10–30% in 0.1 M PBS, pH 7.4). The brains were frozen in 2-methyl butane and kept at −80℃ until sectioning on a cryotome. Coronal sections (25 µm thick) were collected serially, mounted on superfrost glass slides, dried for 2 h at room temperature and stored at −20℃ until staining. Subsequently, the slides were incubated for 60 min at room temperature with one of the following primary antibodies: rabbit anti-glial fibrillary acidic protein (GFAP) (1:500; Dako, Denmark), a specific marker of astrocytes, and rabbit anti-Iba1 (1:100; Abcam, UK), a marker for microglia. Sections were incubated with appropriate peroxidase-linked secondary antibody (1:200; Santa Cruz Biotechnology, USA) and the immunoreaction products were visualized with 3´3-diaminobenzidine (DAB; Dako) according to the manufacturer's instructions. Finally, slides were counterstained with hematoxylin, mounted with Kaiser gel (Sigma-Aldrich) and examined under a Zeiss Axiovert microscope (Carl Zeiss, Germany).
Western blot analysis
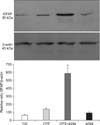
After decapitation, brains were removed from three animals and the cortices were dissected and pooled. The selected tissue was homogenized with a hand-held pestle in sodium dodecyl sulfate (SDS) sample buffer (10 mL/mg) that contained a cocktail of proteinase and phosphatase inhibitors [18]. The electrophoresis samples were heated at 100℃ for 5 min, then loaded onto 10% SDS-polyacrylamide gels with standard Laemmli solutions (BioRad Laboratories, USA). The proteins were electroblotted onto a polyvinylidene difluoride membrane (PVDF, Immobilon-P; Millipore, USA), then placed in a blocking solution that contained Tris-buffered saline with 0.02% Tween (TBS-T) and 5% non-fat dry milk for 1 h. Next, samples were incubated overnight under gentle agitation with primary antibody rabbit anti-GFAP (1:7,000; Dako) and mouse anti-β-actin (1:1,000; Sigma, USA). Bound primary antibodies were detected with horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody (1:5,000; Santa Cruz Biotechnology). Between each step, the immunoblots were rinsed with TBS-T. Immunoreactive bands were visualized on X-ray films (Kodak, USA) using chemiluminescence, after which the optical densities of immunoreactive bands from four independent blots were calculated in the Image Quant program. The densities of GFAP and β-actin immunoreactive bands were quantified with background subtraction. Briefly, squares of identical sizes were drawn around each band to measure density, and the background near that band was subtracted. For each blot, optical densities were normalized against β-actin levels.
Statistical analysis
After verifying a normal distribution in all groups using the Kolmogorov-Smirnov test, the results were presented as the mean ± SEM. The significance of difference between the data obtained for different groups was determined by one way ANOVA using Dunnett's C test. Linear regression analysis was conducted to determine the relationship between the obtained values of parameters using the GraphPad Prism statistical program. A p < 0.05 was considered statistically significant.
Results
The results of our study revealed that CPZ treatment induced different changes in parameters of oxidative stress and antioxidant capacity in forebrain cortex samples of experimental animals. Treatment of rats with AGM (75 mg/kg BW i.p.) alone did not significantly affect any of the measured parameters relative to those that received only saline.
CPZ concentration in the rat forebrain cortex
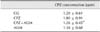
In the CPZ+AGM group, CPZ concentration was significantly decreased in the forebrain cortex (p < 0.05) compared to the CPZ group of animals (Table 1).
Concentrations of parameters of oxidative status in the rat forebrain cortex
The concentration of TBARS in the CPZ group increased significantly 48 h after treatment in the forebrain cortex (p < 0.001) compared to the controls (panel A in Fig. 1). Superoxide anion production increased significantly in the forebrain cortex (p < 0.001) relative to the controls (panel B in Fig. 1). Treatment with AGM applied together with CPZ decreased the O2•− production in the forebrain cortex (p < 0.05) relative to the CPZ group.
In the CPZ group, total GSH content was significantly decreased in the forebrain cortex (p < 0.001) compared to the controls (Fig. 2). Total GSH content significantly increased in both the CPZ+AGM (p < 0.05) and AGM (p < 0.001) groups relative to the CPZ group in the forebrain cortex 48 h after treatment.
Administration of CPZ resulted in decreased total SOD activity (p < 0.001) 48 h after treatment in the forebrain cortex relative to the controls (panel A in Fig. 3). In the CPZ+AGM (p < 0.001) and AGM (p < 0.01) group, total SOD activity increased in the forebrain cortex relative to the CPZ-treated group. In the CPZ+AGM group, CAT activity was significantly incresed in the forebrain cortex (p < 0.05) 48 h after tretment relative to the CPZ group (panel B in Fig. 3).
Administration of CPZ resulted in decreased GPx activity (p < 0.05) in the forebrain cortex relative to the controls (panel A in Fig. 4). In the CPZ+AGM and AGM group, total GPx activity increased (p < 0.001) in the forebrain cortex relative to the CPZ-treated group. In the CPZ group, GR activity was significantly lower (p < 0.05) in the forebrain cortex compared to the control (panel B in Fig. 4) at 48 h. However, GR activity increased significantly (p < 0.01) in both the CPZ+AGM and AGM group in the forebrain cortex relative to CPZ-treated animals at 48 h after tretment.
Correlation Analysis
The results indicate a significant positive correlation (Table 2) between total SOD activity and CAT activity in the forebrain cortex at 48 h after CPZ administration (r = 0.8924, p < 0.01). Additonally, a significant negative correlation between CPZ concentration with both O2•− level (r = −0.8271, p < 0.05) and total GSH concentration (r = −0.9158, p < 0.01) was observed. We also found a negative correlation between GR activity and TBARS concentration in the forebrain cortex 48 h after CPZ treatment (r = −0.8338, p < 0.05) (Table 2).
Immunohistochemistry
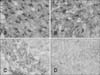
In control rats, we observed fibrous astrocytes with small cell bodies and long, thin processes of individual astrocytes that occupy essentially non-overlapping domains throughout the cortex (panel A in Fig. 5). Conversely, 48 h after CPZ administration hypertrophy of astrocytes and its proceses without substantive loss of individual astrocyte domains was observed (panel B in Fig. 5). However, GFAP labelling demonstrated a mild reaction in the AGM group (panel D in Fig. 5), and prominent staining was present in the CPZ+AGM group (panel C in Fig. 5).
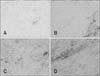
Staining with Iba1 clearly revealed widespread distribution of finely branching microglia and confirmed the essential absence of globoid monocytes or macrophages in the CNS parenchyma of normal rats (panel A in Fig. 6). In the CPZ group of animals, Iba1 staining confirmed a pronounced increase in the number of branched microglia as noted by Iba1 staining in both gray and white matter (panel B in Fig. 6). In the CPZ+AGM group (panel C in Fig. 6) as well as in the AGM group (panel D in Fig. 6), there was a further obvious decrease in the number of cells stained with Iba1 relative to the CPZ group, with much more widespread distribution of Iba1-positive globoid macrophages in the parenchyma.
Western blot analysis
Immunoblot analysis showed that GFAP was present as a single band with a molecular mass of about 50 kDa (Fig. 7). There was a significant increase in GFAP expression followed CPZ+AGM induced brain injury in rats with respect to physiological control and CPZ-induced injury. Moreover, there was no increase in GFAP expression relative to the controls following AGM.
Discussion
Our results revealed that oxidative stress plays an important role in acute CPZ-induced brain injury in rats. The neuroprotective role of AGM could be essential 48 h after CPZ-induced neurotoxicity mediated by oxidative stress.
Application of CPZ after 48 h caused no change in the concentration of the drug in rat brains relative to the control values (Table 1). However, CPZ+AGM significantly reduced the concentration of CPZ in the brain, indicating that the presence of AGM reduced the resorption of deposited CPZ. Application of CPZ+AGM also reduced the distribution of CPZ by compartments. Correlation analysis revealed a positive correlation between the concentration of CPZ and total SOD activity in the forebrain cortex and a negative correlation between the concentration of CPZ and both total GSH and O2•− production in the forebrain cortex 48 h after CPZ administration.
It is well known that oxidation of membrane lipids results in the formation of peroxidation degradation products, which leads to cross-linking reactions of the lipid-lipid and lipid-protein type, thereby rendering the membrane more rigid and less fluid [2229]. The results of the present study showed that brain TBARS concentrations were significantly increased (p < 0.05) in the CPZ-treated group relative to the control group in the forebrain cortex. Oxidative stress was implicated in tissue damage induced by CPZ-treatment [21]. Neurochemical disruption of connectivity between the hippocampus and forebrain cortex, as well as the striatum induced by CPZ administration could provoke retrograde transneuronal damage of the forebrain cortex that could explain the increased TBARS concentration observed in our experiments. The results of the present study are consistent with those of other studies that showed CPZ leads to membrane damage through LPO [2638]. In this study, no change in the concentration of TBARS was observed in the forebrain cortex after CPZ+AGM administration. Moreover, the cortex contains NADPH-dijaforaza-positive neurons, which are resistant to the toxic effects of NO. This could explain the security in this region of the LPO process [10].
Increased O2•− generation in the forebrain cortex after CPZ administration indicates temporal and spatial propagation of the O2•− production process, since it occurs in the brain structure, where there is a suitable biochemical environment for its development. Increased production of O2•− after 48 h indicates a long-term toxic effect of CPZ because it does not establish a redox balance or maintain inadequate AOS. The protective effects of the AGM in our study confirmed that the reduced O2•− production after CPZ+AGM treatment can be explained by its total antioxidant potential.
GSH is a multifunctional, intracellular nonenzymatic antioxidant and the major component of intracellular regulation of the redox state, providing the first line of defense against oxidative injury [27]. GSH is also an important substrate and cofactor in drug metabolism. In our study, a decrease in total GSH concentration relative to the control group was observed 48 h after CPZ administration, which could be explained by the increased activity of γ-glutamyltranspeptidase, an enzyme localized to the outer surface of the cells, where it breaks down extracellular GSH [7]. According to the second explanation, the reduction of total GSH is due to increased LPO in the brain, which is accompanied by a lack of NADPH required for GR activity and transformation of oxidized GSH in the reduced form [9]. The increase of total GSH content in the CPZ+AGM group shows that AGM prevents oxidative damage by increasing the capacity of AOS in the brain tissue of rats. These results show the neuroprotective effects of the AGM under various pathological conditions, including poisoning [3].
The acute administration of CPZ after 48 h leads to a decrease in total SOD activity relative to the control. However, CAT activity was equal to the control values 48 h after CPZ administration in the forebrain cortex. A positive correlation was observed between the activity of SOD and CAT in the forebrain cortex 48 h after CPZ administration. The increase in LPO and increased O2•− production with a decrease of SOD may be associated with an increase in glutamatergic transmission following the chronic blockade of dopamine receptors. Furthermore, our results show that CPZ+AGM reduces O2•− production and increases the activity of the antioxidant enzymes SOD and CAT. A study by Schwartz et al. [37] showed that SOD and CAT prevented glutamate-induced neuronal degeneration.
CPZ reduced the activity of GPx and GR relative to the controls and may be a result of disturbance of redox regulatory mechanisms in the AOS. Reduced activity of GPx and GR in the forebrain cortex shows a loss of total GSH and increased LPO, which is also registered in the cortex after CPZ administration. Our findings show that GR was negatively correlated with the concentration of TBARS in the forebrain cortex 48 h after CPZ administration. Treatment with AGM prevents biological oxidative damage, while administration of CPZ+AGM increases the activity of both GPx and GR, suppresses LPO and prevents the development of oxidative stress.
Astrocytes are integrally involved in metabolic and ionic homeostasis, but also generate neurotrophic factors and provide neurons with precursors necessary to combat oxidative stress [1236]. It is well known that dysfunction of astrocytes in acute brain injury may compromise neuronal metabolism and survival, thereby contributing to neurologic destruction [11]. Damage to the extracellular ionic homeostasis maintained by astrocytes continues to lead to excitotoxic damage [6]. Acute application of CPZ after 48 h led to increased immunoreactivity of glial cells in the rat brain in terms of increasing the number of GFAP-positive cells. These cells are arranged in nonoverlapping spatial domains with pronounced hypertrophy of cell bodies and processes. We also found an increase in the number of GFAP positive cells in the CPZ+AGM group of animals. The administration of AGM in CPZ animals, which induces a glial reaction resembling reactive astrogliosis with a moderate reaction of the GFAP-positive cells, suggests a reparative potential of this molecule in the rat brain.
Based on the electrophoretic profiles of the GFAP molecules in the forebrain cortex, we found clear differences in the level of expression of the protein, which are most pronounced in the CPZ+AGM group of animals 48 h after the treatment. It is well known that astrocytes are important producers of antioxidants in the normal CNS, and astrocytic production of these molecules after brain injury may enhance neuronal survival and protect astrocyte function. Therefore, the increased GFAP expression may be a contraregulatory mechanism to reduce oxidative stress in CPZ+AGM-induced injury.
Reactive astrocytes could have beneficial roles in defense and repair, which include suppression of oxidative radicals and the LPO process [34]. However, under pathological circumstances, glial activation also involves microglia, and their interaction with astrocytes may contribute to secondary nerve-cell damage [8]. Microglia are innate immune cells residing in the CNS. Resting microglia are small cells with long, thin ramified processes. Upon activation of microglia, cell bodies become enlarged and processes become poorly ramified, short, and thick. The brain sections of the control group had few Iba1-positive cells with small cell bodies and long, thin ramified processes. In CPZ-treated animals, the resting microglia became activated, and the number of microglia was significantly increased in both white matter and gray matter of the brain compared with controls. When rats were treated with CPZ+AGM or AGM, there were few Iba1-positive cells in the brain, and these Iba1-positive cells had small cell bodies with long, thin ramified processes.
The results show that AGM reduces brain levels of oxidative stress parameters, improves antioxidant enzymatic and non-enzymatic activities, and reduces reactive astrogliosis and microglia reactivation in CPZ-induced injury, indicating that AGM might exert a protective effect against brain damage.


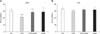
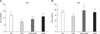

 XML Download
XML Download