

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Dear Editor,
Plasma cell myeloma (PCM) is multifocal neoplastic proliferation of plasma cells originating from the bone marrow (BM) and damaging other organs by production of a monoclonal protein (M protein) in the serum and/or urine [1]. PCM is diagnosed based on comprehensive clinical, morphological, immunological, and radiological evidence [1]. PCM-related end-organ damage typically includes hypercalcemia, renal insufficiency, anemia, and bone lesions (CRAB criteria) [1]. Lytic bone lesions are induced via complex pathophysiology of malignant plasma cells, bone stromal cells, and BM microenvironment [23]. Bone lesions are found in approximately 70% of PCM patients during radiological skeletal surveys [1]. Monoclonal cells can replace normal hematopoietic cells with various patterns of infiltration in BM [1]. Brown tumor (osteitis fibrosa cystica) is a benign bone tumor and metabolic bone disease induced by primary or secondary hyperparathyroidism [4567]. Radiologically, multiple lytic bone lesions are difficult to differentiate from PCM or metastatic cancer; pathologically, it is challenging to discriminate them from giant cell tumors of the bone [45678]. Therefore, calcium and parathyroid hormone (PTH) levels are essential for distinguishing brown tumors from other bone diseases associated with lytic bone lesions. Although increased osteoclasts can be observed in some PCM cases [1], there has been only one report so far of a PCM patient with brown tumor features and severe osteoclastic activity in BM, associated with secondary hyperparathyroidism in response to drug-induced hypocalcemia, and exhibiting serological complete response (CR) [9]. We report the first case of PCM with brown tumor features unrelated to hyperparathyroidism.
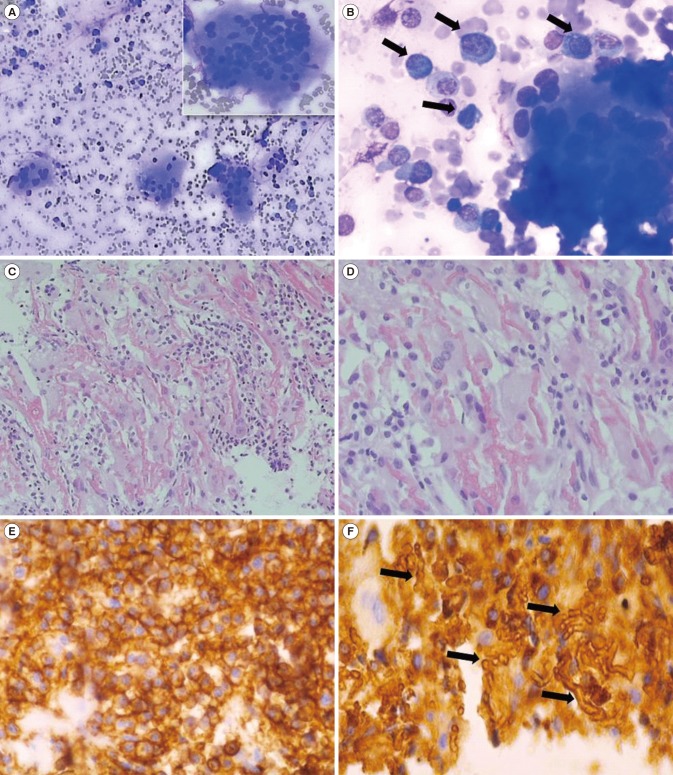
A 50-year-old female presented with right pleuritic chest pain, nausea, and poor oral intake in January 2016. Multiple lytic lesions were detected using X-ray and computed tomography in the skull, spine, humerus, femur, pelvis, and sacrum. Complete blood count showed anemia (Hb 81 g/L). M protein with free kappa chain type in serum and urine was detected by immunofixation electrophoresis and free light chain analysis (free κ/λ ratio, 237.68 and 158.16, respectively). Additionally, calcium level was 3.14 mmol/L, whereas PTH and PTH-related protein (PTHrP) levels were within normal limits (1.06 pmol/L and <1.1 pmol/L, respectively). Creatinine level was 388.96 µmol/L. BM aspirates contained 44% plasma cells with markedly increased clusters of multinucleated osteoclasts (Fig. 1A and 1B) and 28% CD19−CD56+CD138+ plasma cells, which were identified by flow cytometry. The hypercellular BM showed increased malignant plasma cells with tunneling feature, as well as interstitial hemorrhage, woven bone formation by destruction of trabecula, and increased osteoclasts (Fig. 1C and 1D). Monoclonal malignant plasma cells were identified in the BM biopsy by immunohistochemical staining for CD138 and immunoglobulin (Ig) kappa chain; interstitial hemorrhage was observed in the form of crystals after Ig kappa light chain staining (Fig. 1E and 1F).
Serologically, the patient exhibited CR for one year post treatment with a velcade+thalidomide+dexamethasone regimen every three weeks for four courses and autologous peripheral blood stem cell transplantation. However, hemodialysis was needed three times per week for impaired renal function, together with supplemental treatments with pamidronate and salcatonin for hypercalcemia during her survival. PCM relapsed with elevated monoclonal Ig and hepatosplenomegaly with suspicions of plasmacytoma involvement one year following CR. The patient expired in November 2017 with aggravation of liver function and persistent cytopenia despite additional chemotherapy (velcade+dexamethasone+lenalidomide for two three-week cycles).
Our case was distinguished by rare morphological features shared with brown tumors, including an increased number of multinucleated osteoclasts with trabecular destruction and tunneling features on woven bone formation in BM. Increased numbers of osteoclasts secreting growth factors promote monoclonal plasma cell growth and survival, thus altering the balance between bone resorption and formation [3]. Therefore, monoclonal plasma cell proliferation induces tumor progression and osteolysis and suppresses osteoblast function by producing several cytokines such as IL-6, chemokine (C-C motif) ligand 3 (CCL3), and dickkopf-1 (DKK1) [23]. Altered osteoblastic function occurs prior to bone loss in PCM. Malignant plasma cells with brown tumor features involving BM reflect disease aggressiveness, accompanied by severe osteolytic lesions, uncontrolled hypercalcemia, and impaired renal function.
Proliferation of monoclonal plasma cells with brown tumor features is an uncommon BM finding in PCM. Thus, this report might contribute to the differential diagnosis of other osteolytic bone diseases. Pathologically, a preponderance of uncommon BM findings may elucidate the pathophysiology of PCM, could be used to assess disease status, and could provide opportunities
for supplemental treatment of this rare group of patients with PCM.
 XML Download
XML Download