

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Dear Editor,
Hereditary or primary hypertrophic osteoarthropathy (PHO) is an autosomal recessive disorder characterized by excessive proliferation of skin and bone cells at the distal parts of the extremities, resulting in clubbing of fingers and toes, periostitis of long tubular bones, and arthritis [1]. Pathogenic variants in the 15-hydroxyprostaglandin dehydrogenase (HPGD) gene and solute carrier organic anion transporter family member 2A1 gene (SLCO2A1) have been identified in hypertrophic osteoarthropathy, primary, autosomal recessive 1 (PHOAR1; Online Mendelian Inheritance in Man [OMIM] #259100) and hypertrophic osteoarthropathy, primary, autosomal recessive 2 (PHOAR2; OMIM #614441), respectively [23]. Some differences exist between the clinical phenotypes of PHOAR1 and PHOAR2: 1) pachydermia and cutis gyrate are more frequent and severe in PHOAR2 than in PHOAR1; 2) a certain proportion of PHOAR2 exhibits gastrointestinal hemorrhage, which is not observed in PHOAR1; 3) higher urinary prostaglandin E2 (PGE2) levels are observed in PHOAR2 (approximately 10-fold increase) than in PHOAR1 (approximately 4-fold increase); and 4) onset is usually around puberty in PHOAR2, whereas onset is often around birth in PHOAR1 [4]. In Korea, several cases of PHO have been reported, all harboring SLCO2A1 mutations [56]. We report the first case in Korea of a patient with PHO carrying compound heterozygous pathogenic variants in the HPGD gene.
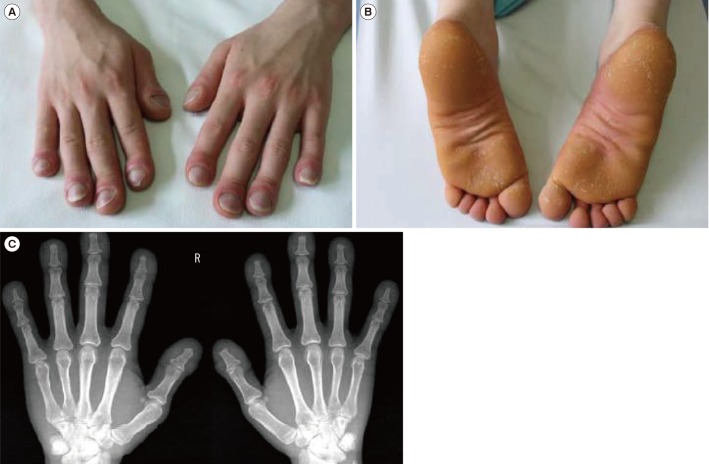
A 21-year-old military soldier was referred to the outpatient clinic of the Armed Forces Capital Hospital, Seongnam, Korea, with a suspected diagnosis of acromegaly in February 2011. He had abnormal thickening of both hands and feet, as well as digital clubbing, which is an unusual finding in patients with acromegaly. The digital clubbing required him to visit a tertiary hospital when he was six years old, but the cause of clubbing was not found. He denied any medical history, including that of gastrointestinal hemorrhage, any affected family members, or a sudden increase in height. Upon physical examination, digital clubbing of the fingers (Fig. 1A) and abnormal thickening of the feet (Fig. 1B) were noted; however, forehead pachydermia was not evident. Laboratory test results were within the reference range (blood chemistry, complete blood cell count, erythrocyte sedimentation rate, and C-reactive protein) as were hormonal test results (total-triiodothyronine, free-thyroxine, thyroid-stimulating hormone, and insulin-like growth factor 1). The chest X-ray showed no abnormalities, whereas the hand X-ray showed soft tissue swelling of the fingers without apparent acroosteolysis (Fig. 1C). No other long bones showed abnormalities. Bone scintigraphy and sella magnetic resonance imaging did not reveal any remarkable findings. Secondary causes of hypertrophic osteoarthropathy were ruled out using echocardiography, pulmonary function test, ultrasonography of the abdomen, and esophagoduodenoscopy. Urinary PGE2 was 2,626 ng, approximately 4-fold higher than the upper normal range (reference range: 400–620 ng/24-hour urine).
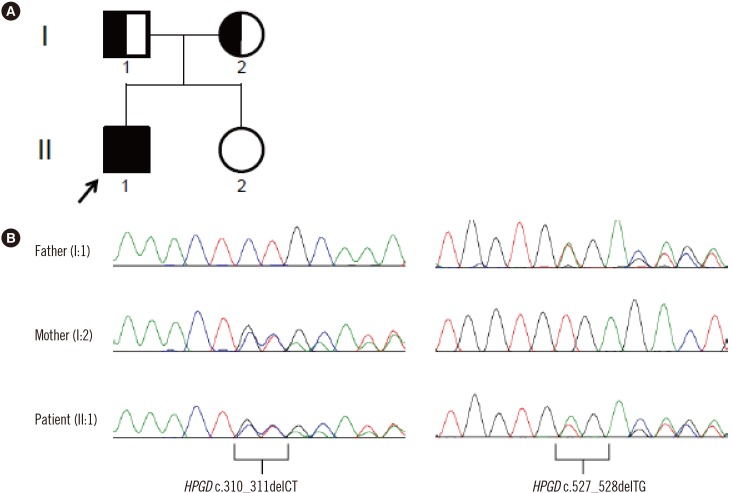
The patient was suspected to have PHOAR1 rather than PHOAR2, and we decided to perform sequence analysis of the HPGD gene. After obtaining informed consent, genomic DNA was extracted from peripheral blood leukocytes of the patient and both parents. All seven coding exons and their flanking intronic regions were amplified and sequenced using an ABI 3730xl genetic analyzer (Applied Biosystems, Foster City, CA, USA) and the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) with primers designed by the authors (available upon request). HPGD sequences were analyzed using Sequencher software (Gene Codes Corp., Ann Arbor, MI, USA) and compared with the reference sequence (NM_000860.5). The patient was compound heterozygous for a 2-bp deletion (NM_000860.5:c.310_311delCT) in exon 3 and a 2-bp deletion (c.527_528delTG) in exon 6, and both parents were heterozygous carriers of each variant (Fig. 2).
The c.310_311delCT variant has been predicted to cause a truncated protein (p.Leu104Alafs*3) and was reported as a pathogenic variant in two pairs of Turkish siblings [78], two Chinese siblings [9], and seven sporadic Chinese patients [9]. The c.527_528delTG variant is predicted to lead to a truncated protein (p.Val176Glufs*11) by Mutalyzer 2.0.28. It is absent from the Genome Aggregation Database (http://gnomad.broadinstitute.org) and the Korean Reference Genome database (http://152.99.75.168/KRGDB/), but it can be classified as a pathogenic variant according to the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [10].
Previously reported patients carrying c.310_311delCT homozygous variants presented common clinical PHOAR1 findings: digital clubbing, periostosis, acroosteolysis, joint swelling, and palmoplantar hyperkeratosis. Tüysüz et al. [7] described that the Turkish boys' father and grandfather also had mild digital clubbing and palmar hyperkeratosis. Carriers of HPGD gene mutations have been known to present some signs of PHO [1]. However, Erken et al. [8] showed that the Turkish siblings' relatives who were carriers did not exhibit signs of PHO. Neither parent of our patient showed any signs of PHO.
Although our patient had abnormal thickening of both hands and feet before school age, he neither experienced hand pain nor exhibited apparent acroosteolysis. Variable clinical phenotypes between PHOAR1 and PHOAR2, as well as within the same disease subtype, have not been completely elucidated; thus, further studies are necessary to elucidate the genotype-phenotype correlation.
 XML Download
XML Download