

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Dear Editor,
Limb-girdle muscular dystrophies type 1D (LGMD1D) is generally known as an adult-onset musculoskeletal disease. However, according to reports, there are cases of onset in children, including Nam, et al.'s study.1 Although most patients with LGMD1D are characterized by an onset in adulthood, some develop at an early age, and as noted, the age of disease onset can vary. Our patients also presented with various clinical manifestations. In our patients, there was no significant evidence of cardiac involvement based on past history, clinical symptoms and signs, findings on physical examination, laboratory data, and chest X-ray. The possibility of subclinical cardiac abnormality in our patients cannot be ruled out completely, however, alhough we believe that our patients did not have cardiac abnormalities at that time.
In addition, serum creatine kinase (CK) levels (our normal range: 35–232 IU/L) are a useful clinical biomarker, but fluctuate and vary, even at low levels in a muscle atrophy state. The serum CK levels of our patients were normal to mildly elevated. The first patient had a CK level of 175 IU/L, and the second patient had levels of 504 IU/L and 673 IU/L on two tests. The last patient had a level of 301 IU/L.
We also agree that the phenotype of LGMD1D may appear more heterogeneous than we thought. This has been confirmed from the paper published by Ruggieri, et al.2 According to the paper, patients with proximal G/F domain mutations (Phe89, Phe91, and Phe93) in DNAJB6 show proximal limbgirdle weakness, whereas patients with distal G/F domain mutations (Pro96 and Phe100) expereience distal leg weakness.
We stated more than nine mutations in the DNAJB6 gene have been reported, including p.Phe89Ile, p.Phe91Ile, pPhe-91Leu, p.Phe93Ile, p.Phe93Leu, p.Pro96Arg, p.Pro96Leu, p.Phe-100Val, and p.Phe100Ile. Recently, two mutations (c.284A>T, p.Asn95Ile, two families; and c.293_295delATG, p.Asp98del, one family) were reported.3 Thus, at least 11 different mutations have been identified so far (Table 1). As next generation sequencing becomes more wildly used, additional novel mutations will be identified.
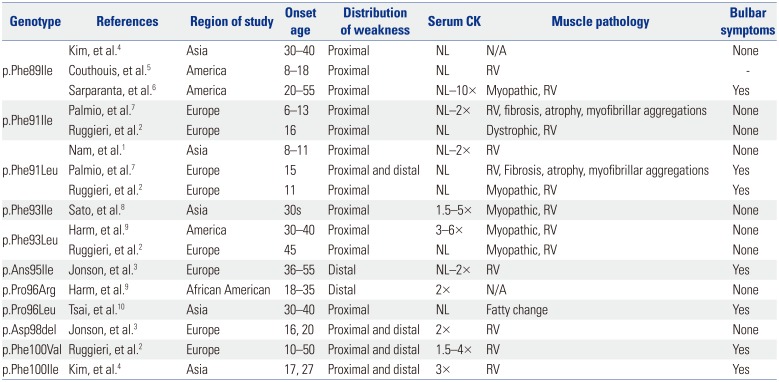
Table 1
Clinical Features and Genetic Findings for DNAJB6 Mutations
| Genotype | References | Region of study | Onset age | Distribution of weakness | Serum CK | Muscle pathology | Bulbar symptoms |
|---|---|---|---|---|---|---|---|
| p.Phe89Ile | Kim, et al.4 | Asia | 30–40 | Proximal | NL | N/A | None |
| Couthouis, et al.5 | America | 8–18 | Proximal | NL | RV | - | |
| Sarparanta, et al.6 | America | 20–55 | Proximal | NL–10× | Myopathic, RV | Yes | |
| p.Phe91Ile | Palmio, et al.7 | Europe | 6–13 | Proximal | NL–2× | RV, fibrosis, atrophy, myofibrillar aggregations | None |
| Ruggieri, et al.2 | Europe | 16 | Proximal | NL | Dystrophic, RV | None | |
| p.Phe91Leu | Nam, et al.1 | Asia | 8–11 | Proximal | NL–2× | RV | None |
| Palmio, et al.7 | Europe | 15 | Proximal and distal | NL | RV, Fibrosis, atrophy, myofibrillar aggregations | Yes | |
| Ruggieri, et al.2 | Europe | 11 | Proximal | NL | Myopathic, RV | Yes | |
| p.Phe93Ile | Sato, et al.8 | Asia | 30s | Proximal | 1.5–5× | Myopathic, RV | None |
| p.Phe93Leu | Harm, et al.9 | America | 30–40 | Proximal | 3–6× | Myopathic, RV | None |
| Ruggieri, et al.2 | Europe | 45 | Proximal | NL | Myopathic, RV | None | |
| p.Ans95Ile | Jonson, et al.3 | Europe | 36–55 | Distal | NL–2× | RV | Yes |
| p.Pro96Arg | Harm, et al.9 | African American | 18–35 | Distal | 2× | N/A | None |
| p.Pro96Leu | Tsai, et al.10 | Asia | 30–40 | Proximal | NL | Fatty change | Yes |
| p.Asp98del | Jonson, et al.3 | Europe | 16, 20 | Proximal and distal | 2× | RV | None |
| p.Phe100Val | Ruggieri, et al.2 | Europe | 10–50 | Proximal and distal | 1.5–4× | RV | Yes |
| p.Phe100Ile | Kim, et al.4 | Asia | 17, 27 | Proximal and distal | 3× | RV | Yes |
![]()
 XML Download
XML Download