

 PDF
PDF ePub
ePub Citation
Citation Print
Print



We read with interest the article by Kim, et al.1 concerning 3 patients with autosomal dominant limb girdle muscular dystrophy type 1D (LGMD1D) due to mutations p.Phe89Ile and p.Phe100Ile in DNAJB6. We have the following comments and concerns.
If patients with LGMD1D exhibit symptoms in childhood, it is not justified to call LGMD1D a disorder only with adult onset. At least, a rare early onset and the more common late onset phenotype should be designated. Not only did Patient-2 experience muscle problems in childhood,1 but also Ruggieri, et al.2 reported a patient with childhood onset. Another LGMD1D patient with childhood onset (8 years) was described by Suarez-Cedeno, et al.3 Finally, Nam's index case had early onset at age 8 years.4
Additionally, if a patient does not complain about cardiac symptoms, this does not necessarily mean that he has no cardiac involvement. Cardiac disease can be subclinical, and thus, all patients with myopathy need to undergo prospective cardiac investigations for cardiac involvement. Cardiac involvement frequently comprises cardiomyopathy of any type (noncompaction, heart failure, or arrhythmias). Accordingly, I wonder whether there were any abnormalities detected on long-term ECG echocardiography or cardiac MRI.
Typically, for most neuromuscular disorders, creatine-kinase (CK) values fluctuate. Thus, it is essential to review previous laboratory investigations to see if CK was elevated prior to diagnosis. I would be interested to know to what degrees did CK values fluctuate in the three presented patients over time. Moreover, a serum CK value of 175 U/L, as detected in Patient 1, would be abnormal in our laboratory. Provision of reference limits would be helpful.
Though rare, it should be mentioned that LGMD1D may have an onset with distal muscle weakness (Table 1), as has been reported by Ruggieri, et al.2 There are also patients who generally have distally pronounced muscle weakness (Table 1).5 Distal predominance has also been reported in the three affected members of family 2 reported by Harms, et al.6 (Table 1). Two of the patients described by Kim, et al.1 had distal predominance as well (Table 1). Generally, the phenotype of LGMD1D seems to be more heterogeneous than initially assumed.
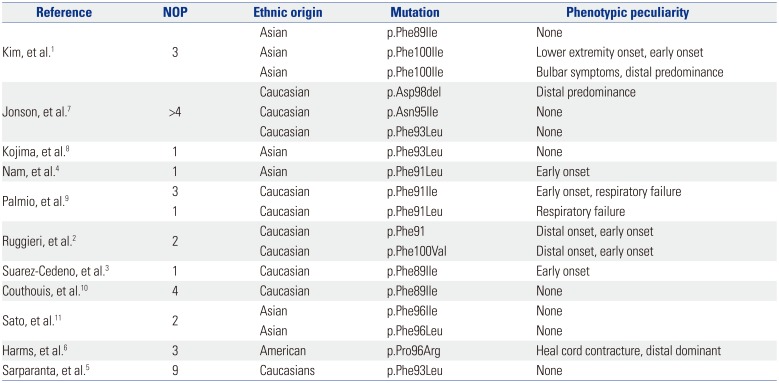
Table 1
Genetic Background of Patients with Limb Girdle Muscular Dystrophy Type 1D
| Reference | NOP | Ethnic origin | Mutation | Phenotypic peculiarity |
|---|---|---|---|---|
| Kim, et al.1 | 3 | Asian | p.Phe89Ile | None |
| Asian | p.Phe100Ile | Lower extremity onset, early onset | ||
| Asian | p.Phe100Ile | Bulbar symptoms, distal predominance | ||
| Jonson, et al.7 | >4 | Caucasian | p.Asp98del | Distal predominance |
| Caucasian | p.Asn95Ile | None | ||
| Caucasian | p.Phe93Leu | None | ||
| Kojima, et al.8 | 1 | Asian | p.Phe93Leu | None |
| Nam, et al.4 | 1 | Asian | p.Phe91Leu | Early onset |
| Palmio, et al.9 | 3 | Caucasian | p.Phe91Ile | Early onset, respiratory failure |
| 1 | Caucasian | p.Phe91Leu | Respiratory failure | |
| Ruggieri, et al.2 | 2 | Caucasian | p.Phe91 | Distal onset, early onset |
| Caucasian | p.Phe100Val | Distal onset, early onset | ||
| Suarez-Cedeno, et al.3 | 1 | Caucasian | p.Phe89Ile | Early onset |
| Couthouis, et al.10 | 4 | Caucasian | p.Phe89Ile | None |
| Sato, et al.11 | 2 | Asian | p.Phe96Ile | None |
| Asian | p.Phe96Leu | None | ||
| Harms, et al.6 | 3 | American | p.Pro96Arg | Heal cord contracture, distal dominant |
| Sarparanta, et al.5 | 9 | Caucasians | p.Phe93Leu | None |
![]()
Finally, we do not agree with the statement that only nine different mutations have been detected in DNAJB6 so far.1 According to Table 1, at least 10 different mutations have been reported thus far (Table 1).
Overall, this interesting study could be more meaningful by distinguishing between early and late onset LGMD1D, by differentiating distal and proximal predominant affection, by prospective studies of the heart, eyes, ears, gastrointestinal tract, and kidneys to exclude multisystem involvement and by reviewing previously determined CK values.
 XML Download
XML Download