

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Gorlin-Goltz syndrome, also known as basal cell nevus syndrome (BCNS), is a rare autosomal-dominant inherited disorder with complete penetrance and variable expressivity. First described in 1960 by Dr. Gorlin and Dr. Goltz1, it is characterized by the development of multiple basal cell carcinomas (BCCs) in early adulthood along with keratocystic odontogenic tumors (KCOTs) of the jaw, palmoplantar pits, intracranial ectopic calcifications and bony deformities. The most common mutations are heterozygous germline mutations of patched 1 (PTCH1)23, PTCH24 and, less frequently, suppressor of fused gene (SUFU) genes5. These genes act as tumor suppressor genes and participate in the sonic hedgehog pathway.
The hedgehog pathway inhibitors (HPIs), including vismodegib and sonidegib, block the smoothened protein receptor and downstream pathway. Vismodegib was approved by the United States Food and Drug Administration in 2012. The grade of recommendation was 1A for locally advanced or metastatic BCC and 2A for BCNS6. Here, we introduced a strategic vismodegib treatment for tumor shrinkage. Additionally, we evaluated its efficacy and adverse effects. It was the first patient with BCNS to receive vismodegib treatment in Taiwan.
Go to : 
CASE REPORT
We received the patient's consent form about publishing all photographic materials. We had that confirmation in March, 2017 for previous submit for Dermatologica Sinica. A 53-year-old man came to our clinic with lesions on his scalp that had been slowly growing over the previous 2 years. Physical examination detected many black plaques of various sizes, with rolled borders and atrophic and/or ulcerative centers. They were distributed mainly on the head and neck and were scattered over the arms and legs (Fig. 1A). The largest plaque was on the right popliteal fossae (Fig. 2A, top left). Review of the patient's medical history revealed that a large radicular cyst on the jaw had been excised in 1994 and a congenital cleft palate repaired in childhood. He also had a cataract and a detached retina. In addition, he had mild mental retardation. Other physical findings included prominent macrocephaly, mandibular prognathism, multiple palmoplantar pits and bony deformities such as kyphoscoliosis and pectus excavatum. His skull X-ray showed calcification of the falx cerebri (Fig. 1B). Whole-body computed tomography detected neither obvious lymphomesenteric cysts nor lymphadenopathy. As noted above, a diagnosis of Gorlin syndrome (GS) was made.
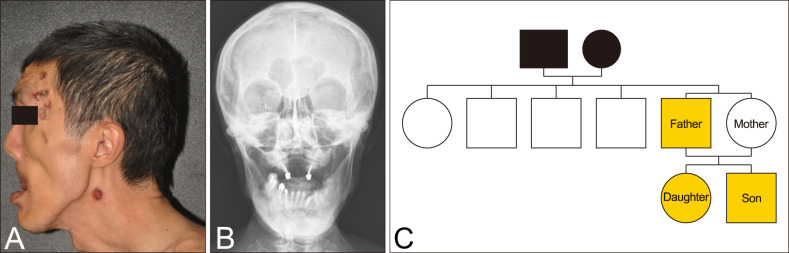 | Fig. 1(A) Many black plaques of various sizes, with rolled borders and atrophic and/or ulcerative centers mainly on the head and neck and prominent frontal bossing. (B) The skull anteroposterior view demonstrated macrocephaly, calcification of the falx cerebri, cleft palate and one osteolytic changes in the mandible secondary to odontogenic cyst. (C) The family pedigree of our cases.
|
According to the patient, neither his parents nor his siblings exhibited the clinical features of GS (Fig. 1C). However, both of his 18-year-old daughter and 15-year-old son fulfilled the diagnostic criteria of BCNS. They had multiple palmar pits, calcification of the falx cerebri, macrocephaly, hypertelorism and multiple KCOTs over the mandible area, and they had undergone several cystectomies. When the children were seen in our clinic, they did not have prominent cutaneous nevoid lesions.
Additionally, the patient's daughter had medulloblastoma with hydrocephalus, which had been diagnosed at 5 months old. She had completed a full course of chemotherapy, with the sequela of vision loss in the right eye; she also had mild mental retardation. A chest X-ray showed scoliosis of the thoracolumbar spine and sprayed ribs. Her younger brother had ventriculomegaly with bilateral subdural collection and had been diagnosed with attention deficit disorder (ADD) by a psychiatrist. At the age of 1 year, he had undergone bilateral herniorrhaphy and orchidopexy. His physical examination in our clinic showed prominent scoliosis and pectus excavatum. Chest radiography revealed that the right third rib was forked. The boy's clinical features are summarized in Table 1.
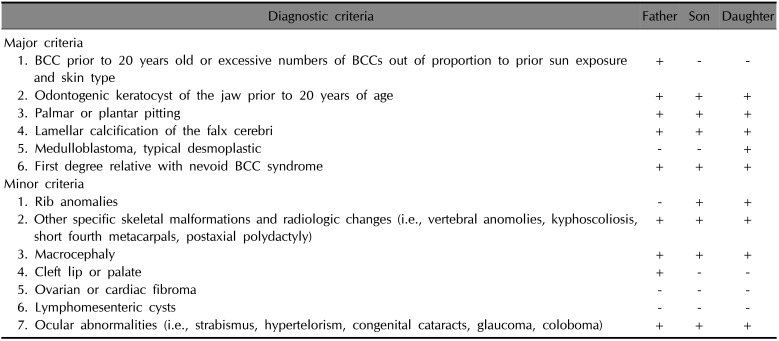
Table 1
Diagnostic criteria of Gorlin-Goltz syndrome and clinical manifestations in our patients
The diagnosis of Gorlin syndrome was based on (1) one major criterion and molecular confirmation; (2) two major criteria; or (3) one major and two minor criteria. BCC: basal cell carcinoma. Adapted from the criteria outlined by the 2011 Consensus Statement from the First International Colloquium on Basal Cell Nevus Syndrome12.
![]()
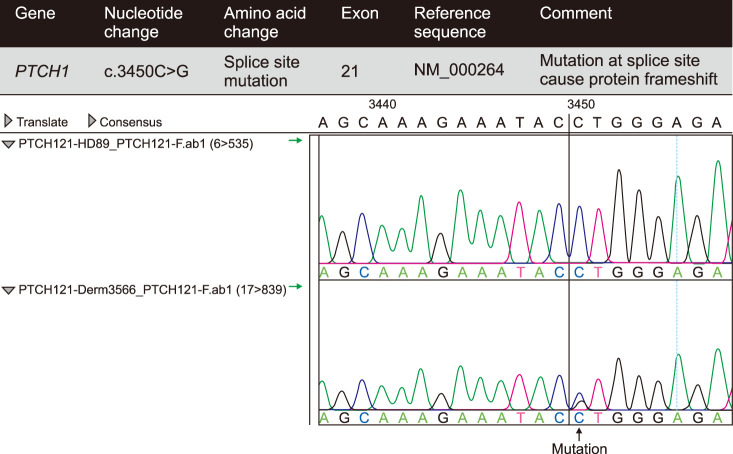
We collected the three patients' genomic DNA from the peripheral blood and used Sanger sequencing to screen the PTCH1 gene for mutations. We used 24 pairs of primers previously reported for the amplification of the 23 exons of the PTCH1 gene7. The mutation of PTCH1 in our cases was identified on the 21st exon and nucleotide transition at 3450 and was a C-to-G transition (Fig. 3). This belongs to the novel mutation site of PTCH1 according to the Human Gene Mutation Database and the NCBI dbSNP website (https://www.ncbi.nlm.nih.gov/snp).
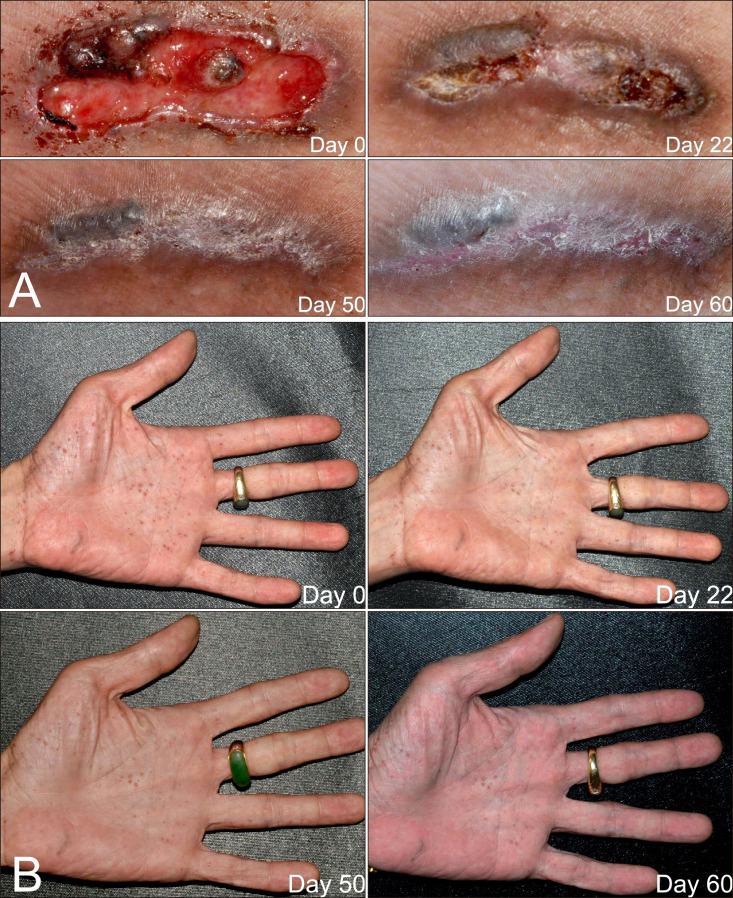
To make the lesions resectable, the father was treated with vismodegib 150 mg per day from August 16, 2016 to October 16, 2016. It was discontinued unilaterally by the patient from August 23 to August 28 because of a runny nose and productive cough. General malaise and soreness occurred a few days after starting to take the medication and constipation developed approximately 2 weeks later. The most intolerable side effect was fatigue, which persisted until the treatment was discontinued. Other side effects included arthralgia and muscle pain. Notably, the patient had a body weight loss of about 7 kilograms and frequent upper airway infections. He had two episodes of common cold, which was more frequent than usual for him, although he was a heavy smoker. Ulceration of the lesions improved, and they gradually regressed (Fig. 2A). In addition, the palmoplantar pitting was considerably ameliorated (Fig. 2B). Electrolyte levels and results of liver and renal function tests were all within normal limits, except for the presence of mild normocytic anemia (hemoglobin 13.3 g/dl). The vismodegib treatment was discontinued when resection of the lesions was feasible.
For long-term management, the patient received total excisions, curettage and electrodessication for the residual lesions every 3 months. Pathological analysis of the largest lesion identified multicentric superficial basal cell carcinoma. New, small lesions appeared gradually but were easily managed by curettage.
Go to :
DISCUSSION
The genetic mutations of GS are commonly PTCH1 on chromosome 9 (9q22.3~q31), PTCH2 on chromosome 1 (1p32.1~32.3) and, rarely, SUFU on chromosome 10q24.328.
The mutation analysis in our patients showed one copy of mutation at PTCH1c.3450C>G. The site of mutation was previously reported by Jayaraman et al.9, but in that case it was a C-to-T transition that caused splice site protein changes in the 1150 amino acids in the extracellular domain. Our cases were C-to-G transitions, which could be compatible with GS.
Clinically, the most striking features were those of GS, which causes both physical and mental impairment. Both the father and the daughter had mild mental retardation, and the son had ADD. Mentally challenging conditions would limit their employment opportunities and require more community resources. Notably, before they were finally diagnosed with GS, they had undergone serial surgeries for their KCOTs. Additionally, the father had received numerous cryotherapy treatments for his palmar pittings, which were misdiagnosed as warts before he noticed his BCCs. Therefore, we should keep in mind that some GS features might have an earlier onset than that of BCCs. We should be more vigilant in order to promptly diagnose individuals who present with GS features.
HPIs target the Smoothened protein and suppress carcinogenesis. They provided alternative approaches to controlling BCCs in patients who are not suitable candidates for radiation therapy or surgery.
Fig. 2A shows significant improvement of the ulceration and gradual regression of the BCCs. Treatment for only 56 days achieved nearly total healing of the ulcers. No new lesions were observed during the entire treatment period. Because vismodegib is both expensive and intolerable by many patients, we present a better way to control BCCs in GS. The concept is similar to that of neoadjuvant therapy in chemotherapy. A tolerable short treatment course could make lesions more accessible for excision.
The adverse reactions most commonly reported (≥10%) are muscle spasms, alopecia, dysgeusia, weight loss, fatigue, nausea, diarrhea, decreased appetite, constipation, arthralgias, vomiting, and ageusia. Tang et al.10 reported a dropout rate of 54% owing to adverse effects. In a study by Yang and Dinehart11, an intermittent dosage was applied, which enhanced compliance. Dréno et al.12 compared two intermittent dosing regimens: treatment group A received 150 mg oral vismodegib per day for 12 weeks and then three rounds of 8 weeks of placebo daily followed by 12 weeks of 150 mg vismodegib daily; treatment group B received 150 mg oral vismodegib per day for 24 weeks and then three rounds of 8 weeks of placebo daily followed by 8 weeks of 150 mg vismodegib daily. The mean number of BCCs at week 73 was reduced more from baseline in treatment group A. Group B had more treatment-related adverse events of grade 3 or worse, higher levels of blood creatine phosphokinase and a higher dropout rate compared with treatment group A.
Our patient received the standard dosage of 150 mg vismodegib once daily for a continuous 56-day treatment. Frequent common colds were not reported as common adverse effects in the previous studies, but our patient was a heavy smoker. It is not clear whether the drug or the patient's ethnicity would interfere with immunity or the drug's efficacy, respectively. Further investigation is needed. Clinically, in individuals treated with HPIs, we should monitor liver function tests, electrolyte levels, bone density and lipids in premenopausal women and cardiovascular status (grade of recommendation 2B)13. Only mild anemia was found in our case.
In summary, a strategic short-term vismodegib treatment course for unresectable BCCs could be a better way to manage patients with GS. This might enhance both compliance and cosmetic outcomes. More cases should be studied to obtain more detailed evaluations.
Go to :
 XML Download
XML Download