

 PDF
PDF ePub
ePub Citation
Citation Print
Print



I. Introduction
Dyke-Davidoff-Masson syndrome (DDMS) is a rare condition that is characterized by hemi-cerebral atrophy, calvarial thickening, hyperpneumatization of the frontal sinuses, and facial asymmetry1. It is usually secondary to trauma to the developing brain in utero or in early childhood2. Trauma to the developing brain results in loss of neurons that comprise the growth of the developing brain, leading to mental retardation, seizures, and learning disability3. Presentation of classically observed characteristics depends on the age of trauma to the developing brain2. Childhood presentation may differ from that of adult if the trauma to the brain presents later in life. The majority of cases reported in the literature showed that this condition is predominant in male patients and in the left hemisphere1. The etiology of DDMS is still controversial, but trauma, infection, intracranial bleeding, congenital vascular anomalies, and peritoneal hypoxia are the leading etiology of this syndrome. Since 1933, less than 100 cases of DDMS have been reported, but none of the cases to date have presented with oral manifestations (clinical and radiographic). The present case describes a 12-year-old male child with classical oral (clinical and radiographical) manifestations of DDMS that contributed to diagnosing the acquired type of DDMS.
II. Case Report
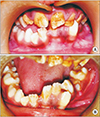
A 12-year-old boy reported to the department of oral medicine and radiology, with chief complaints of difficulty in mastication due to pain in the mandibular left premolar region that had persisted for one week. The patient's past history revealed an uneventful antenatal period and no complications in the post-natal period, but delayed developmental milestones were noted. There was no history of seizures or hemiparesis. The patient demonstrated slurred speech, delayed language learning, and mental retardation. Extremities were normal with both hands and feet of equal size. Facial asymmetry was noticeable, with an enlarged skull (parietal bone) on the left side. The facial skin on the same side showed generalized mild macular brownish diffuse pigmentation similar to café-au-late pigmentation.(Fig. 1. A, 1. B) Eye examination showed decreased vision in the left eye and a lipodermoid, for which surgical correction was advised.(Fig. 1. C) Intraoral examination revealed multiple over-retained right side maxillary and mandibular primary teeth that led to severe malocclusion with an open bite. The permanent maxillary anterior teeth were hypoplastic.(Fig. 2. A) The mandibular left second premolar was erupting bucally.(Fig. 2. B) The primary mandibular left second molar was mobile and slightly tender to vertical percussion. The patient underwent a panoramic radiograph and computed tomography (CT) scan.
The panoramic radiograph showed over-retained primary right-sided mandibular and maxillary canines and first and second molars. Delayed eruption of the permanent maxillary and mandibular canines, first and second premolars, and second molars on the right side was observed. The mandibular left second premolar was obliquely placed. Complete root and 3/4th crown resorption was present with the mandibular primary second molar, and the permanent maxillary and mandibular first molars showed taurodontism. The tooth buds of the permanent maxillary right third molar, bilateral permanent mandibular third molars, and maxillary left second premolar were missing.(Fig. 3) Overall, there was delayed eruption of the permanent teeth on the right side compared to the left, with the exception of the permanent first molars on the right side, which erupted normally in parallel with those on the left side.
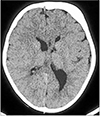
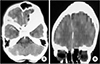
The CT study of the brain (plain and contrast) showed mild atrophy of the left cerebral hemisphere in the form of left lateral ventricle dilatation, sulcogyral space prominence, and cystic parenchymal changes. There was mild shifting of the midline structures toward the left.(Fig. 4) Other characteristic findings on the left side included frontal sinus enlargement, calvarial thickening of the frontal bone and parietal bone, thickening of the squamous part of the temporal bone, and mild elevation of the left orbital roof.(Fig. 4, 5) A diagnosis of DDMS was made based on the characteristic imaging findings.
Considering the complexity of the case, only symptomatic treatment was planned by the pediatric dentist, which included extraction of the deciduous mandibular second molar and follow-up for the erupting mandibular second premolar. However, the patient was lost to follow-up and further treatment.
III. Discussion
Dyke-Davidoff-Masson syndrome was first described by Dyke, Davidoff, and Masson1 in 1933; it is a rare syndrome with characteristic imaging findings. Two types of DDMS have been identified, congenital and acquired. The congenital type involves utero vascular occlusion of the middle cerebral artery, unilateral cerebral arterial circulation anomalies, coarctation of the mid-aortic arch, or infections, and the patient becomes symptomatic in infancy. The acquired type results from various causes such as birth asphyxia, prolonged febrile seizures, trauma, tumor, infection, ischemia, and hemorrhage45. Thus, the age of presentation depends on the time of vascular occlusion of the middle cerebral vascular territory. This injury to the brain leads to cerebral hemiatrophy, which is associated with hypoperfusion and decreased metabolic activity of the affected cerebrum67. The cerebral atrophy is followed by compensatory changes in the adjacent areas only if brain damage occurs before three years of age8. Thus, overlying structures in the brain grow inward, resulting in ipsilateral calvarial thickness, hyperneumatization of the ipsilateral paranasal sinuses, enlargement of mastoid cells, and an elevated orbital roof. Similar findings to this case have been reported, which suggests that the case could be a congenital variant of DDMS. These characteristics are very well documented and can be identified radiographically by CT imaging. Other characteristic changes, which could also be seen in these patients, are elevation of the petrous ridge and sphenoid wing, hypoplasia of the anterior or middle cranial fossa, and shifting of the midline structures toward the atrophic hemisphere910. Such classical radiographic features have been reported in most DDMS cases111213. Further, magnetic resonance imaging helps to clarify the extent of cerebral parenchymal involvement14. In addition to these characteristic imaging findings, clinical findings of seizures, hemiparesis, and mental retardation are also characteristic of a DDMS diagnosis1112.
To date, reported DDMS cases have included patients as young as 18 months to as old as 42 years, and almost all of the cases have reported seizures and hemiparesis of varying intensity11121315. Because the severity of clinical features depends on extent of brain injury, in this case, the cerebral hemiatrophy was mild, and mental retardation, learning disability, speech or language disorders, and facial asymmetry were the only presenting features. While hemiparesis, seizures, and psychiatric disorders were absent, it is possible that these characteristics could manifest later in adolescence. Seizures usually postdate hemiparesis by months or years4, and the prognosis of these patients is considered to be better if the onset of hemiparesis is after 2 years of age and in the absence of seizures16. Thus, the prognosis in this present case could be considered better than other reported cases.
Moreover, the present case manifested with other findings that have not been reported in other DDMS cases: café-au-lait pigmentations on the facial skin of the affected side and lipodermoid of the left eye. Café-au-lait pigmentations are typically seen in neurofibromatosis type I, Albright syndrome, and Silver-Russell syndrome as well as other conditions17, whereas ocular lipodermoids are choristomas that contain adipose tissue, are commonly associated with Goldenhar syndrome18, and rarely require surgery, only if the condition interferes with vision.
Another interesting finding in this patient was delayed eruption of both maxillary and mandibular permanent teeth on the right side compared to left side. Dutta et al.11 and Sethi et al.19 reported shortening of the right hand and foot in patients with left cerebral hemiatrophy. This correlates with the dental findings of the present case, even though the extremities did not demonstrate any variations. In this case, occlusal imbalance and the mobile mandibular primary second molar lead to difficulty in mastication.
This is the first case report of the DDMS syndrome identified by dental findings. In most DDMS patients, seizures and hemiparesis are significant findings that require identification and treatment on a priority basis. Therefore, it is possible that the oral cavity is neglected or given lower priority. This could provide an explanation for the absence of DDMS cases identified via classical dental findings. In the present case, because seizures and hemiparesis were absent, the patient's parent was more concerned about the oral issue. Early identification of the dental problems could prevent already compromised patients from further dental complications. In addition, the parents of these patients could be educated and advised of preventive measures, which include proper oral hygiene, pit and fissure sealants, fluoride application, and regular dental check-ups. As these patients are on long standing anticonvulsants, there is a possibility for gingival hyperplasia, which could lead to periodontitis and early exfoliation of teeth without adequate care. There is no specific treatment for delayed eruption of teeth, but proper counselling and assurance might help prevent further aggravation of psychiatric problems in these patients.
In regard to the delayed eruption of teeth, many conditions have been reported to date; however, unilateral delayed eruption has not been reported. In addition, taurodontism in all of the permanent first molars could be an isolated finding or a component of the syndrome. Because this has not been observed in other cases, the correlation may be difficult to diagnose. Taurodontism has been reported commonly in amelogenesis imperfecta, Down syndrome, Klienfelter syndrome, and Orofaciodigital syndrome20. Of note in this case, in spite of multiple delayed eruptions of permanent teeth on the right side compared to the left, the permanent first molars on the right side erupted normally. In addition, the permanent incisors on the right side were mildly delayed in eruption compared to the left side.
The initiation of calcification of the first permanent molar begins at birth, and initiation of the permanent incisors occurs at approximately six months to one year of age, with the other teeth developing later. This indicates that, in the present case, the time of injury to the brain on the left side could possibly be between the ages of six months to one year because the permanent first molars were relatively unaffected. Observation from a panoramic radiograph adds information to the CT observations that the brain injury, rather than being congenital, is actually acquired and occurred before the age of one year. The oral (clinical and radiographical) findings thus serve as important adjuvant observations that can guide interpretation of the possible time of injury to the brain and, hence, the type of DDMS.
A differential diagnosis of DDMS includes Sturge-Weber syndrome, Rasmussen encephalitis, hemiconvulsionhemiplegia-epilepsy (HHE), Silver-Russell syndrome, basal ganglia germinoma, Fishman syndrome, and linear nevus syndrome2910. The Sturge-Weber syndrome classically includes port-wine facial nevus, leptomeningeal angioma, and intracranial tram track calcification in addition to cerebral atrophy. Rasmussen encephalitis is an immune-mediated brain disorder that affects children between 6 and 8 years of age, leading to unilateral hemispheric atrophy, intractable focal epilepsy, and progressive neurological dysfunction. This condition can be very well differentiated on the basis of imaging features, which include unihemispheric focal cortical atrophy and ipsilateral head of the caudate nucleus, without any calvarial changes21. HHE is characterized by unilateral prolonged clonic seizures followed by the development of hemiplegia in younger children22. Patients with Silver-Russell syndrome have a normal head circumference and normal intelligence but poor growth, clinodactyly, hemi-hypertrophy, and a characteristic facial phenotype (triangular face, small pointed chin, broad forehead, and a thin wide mouth)23. Basal ganglia germinoma is a rare tumor of the brain, associated with progressive hemiparesis and cerebral hemiatrophy. However, imaging reveals cystic areas, focal hemorrhages, and mild surrounding edema along with calvarial changes24. Fishman syndrome or encephalocraniocutaneous lipomatosis is a rare neurocutaneous syndrome that includes unilateral cranial lipoma with lipodermoid of the eye, which typically presents with seizures. Neuroimaging, however, shows a calcified cortex and hemiatrophy25. Linear nevus syndrome could be differentiated by typical facial nevus and unilateral ventricular dilatation that resembles cerebral hemiatrophy, in addition to recurrent seizures and mental retardation26. Treatment options in symptomatic patients include anticonvultion drugs for control of seizures, physiotherapy for hemiparesis, speech therapy for speech defects, and psychiatric counselling and medications, if required. For intractable and uncontrolled seizures and hemiplegia, hemisperectomy is the treatment of choice and has a reported 85% success rate16.
In conclusion, this study reports a possible mild case of DDMS with absence of hemiparesis and seizures. Classical oral manifestations are reported and emphasize the need for dental referrals for such patients to provide preventive oral care, in addition to early detection of malocclusion and other oral problems to provide prompt treatment and avoid further oral complications in an already compromised patient. These oral observations can also be useful in diagnosing this type of DDMS.


 XML Download
XML Download