

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Breast cancer is the most common malignant tumor diagnosed among women worldwide. Despite continued improvement in treatment and earlier detection, breast cancer is the second leading cause of death from cancer in women. As per survey, there are 1.7 million new cases and 521,900 deaths in 2012 [1]. Reportedly, metastasis was found more responsible for the vast majority of cancer patient deaths. Breast cancer cell metastasizes to several organs, however bone is the most preferential metastatic target of breast cancer cells.
To support cancer cell growth, progression and metastasis, tumor cells communicate with other surrounding cells via producing cytokines. Ones of such tumor-derived cytokines influencing progression and metastasis process of breast cancer cells are interleukin (IL)-8 and IL-11. These cytokines are known to possess multiple effects on primary tumor and bone metastasis microenvironment. Studies has elucidated that IL-8 can promote breast tumor cells growth [23], migration, invasion [23] and angiogenesis [45]. Similar to IL-8, IL-11 has also been demonstrated to support breast tumor growth via non-autonomous effects [6]. IL-8 is able to promote early micro-metastatic colonies formation in bone [7]. While, transcription level of IL-11 in patients with breast cancer was found associated with subsequent development of bone metastasis [8]. Moreover, both the cytokines are known to act as osteolytic factors by supporting osteoclastogenesis [91011]. Taken together, IL-8 and IL-11 can be considered for playing important roles in breast cancer progression and osteolytic bone metastasis.
Lysophosphatidic acid (LPA) has been known as a bioactive phospholipid mainly derived from active platelets and several other cell types. Recent study suggested that Autotaxin (ATX)-LPA axis was responsible for bone metastasis by breast cancer cells [12]. As growth factor, LPA has diverse biological activities such as regulation of cell function, proliferation, differentiation, migration and survival in many cell types [13]. The biological action of LPA is mediated by a family of G protein-coupled receptors (GPCRs) called endothelial cell differentiation gene (EDG) family. Breast cancer cells express almost three LPA receptors (LPARs) EDG-2 (LPAR1), EDG-4 (LPAR2) and EDG-7 (LPAR3) which have high affinity for LPA [14]. It has recently emerged that LPA is involved in cancer metastasis and osteolytic bone metastasis although its mechanism of action is poorly understood. Previous studies have shown that LPA induces breast cancer cells proliferation and migration involving signaling pathways like Phosphoinositide 3-kinase (PI3K), mitogen-activated protein kinase (MAPK) and β-catenin [151617]. In addition, LPA may initiate metastasis process via inductive effect on angiogenesis which is mediated by IL-8 and vascular endothelial growth factor (VEGF) [1819]. Several evidences in animal models also suggest the role of LPA in bone metastasis of breast cancer cells [1220].
Therefore, this study was designed to investigate whether LPA can regulate osteolytic factors IL-8 and IL-11 secretion from breast cancer cells and whether can influence breast cancer cells-mediated bone osteoclastogensis via these secretory factors.
METHODS
Reagents and antibodies
Recombinant mouse receptor activator of nuclear factor kappaβ ligand (RANKL) was purchased from BioVision Inc. (Milpitas, CA, US). Oleoyl-L-α-lysophosphatidic acid sodium salt (LPA) was from Sigma (US). LPA receptor-1/2/3inhibitor Ki16425 was bought from Santa Cruz Biotechnology (CA, US). 3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide (MTT) were purchased from Amresco (Albany, NY, US). PD98059, SB203580, SP600125, LY294002, BAY-11-7082, Go6976 and GF109203X were purchased from Tocris Bioscience (Bristol, UK), Y27632 from Cayman Chemical (Ann Arbor, MI, US). Phospho-(pan) PKC (Ser660) antibody was from Cell Signaling Technology (Boston, US). Anti-β-actin was obtained from Santa Cruz Biotechnology. Peroxidase-AffiniPure goat anti-rabbit IgG (H+L) and peroxidase-AffiniPure goat anti-mouse IgG (H+L) were supplied from Jackson ImmunoResearch (Baltimore, US).
Cell culture
Human breast cancer cell line MDA-MB-231 (American Type Culture Collection, ATCC, HTB-26) and MDA-MB-468 (ATCC, HTB-132) were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (all from Gibco, Invitrogen). Cells were maintained at 37℃ and 5% CO2 in a humidified atmosphere. Cells were starved in serum-free media for 12 h prior to LPA treatment.
Murine macrophage cell line RAW264.7 (ATCC, TIB-71) was maintained in an undifferentiated state by culturing in DMEM containing 10% FBS and 1% penicillin-streptomycin at confluences of less than 80%. To induce osteoclastogenesis, RAW264.7 cells were seeded at density of 2×103 cells/cm2 of culture plate for overnight. Then cells were shifted into differentiation media containing Minimum Essential Medium Eagle-Alpha Modification (Alpha-MEM) (Gibco) supplemented with 10% FBS, 1% penicillin-streptomycin and 30 ng/ml RANKL.
Collection of conditioned media (CM)
MDA-MB-231 and MDA-MB-468 cells were plated at a density of 1.0×106 cells/60 mm dish in complete DMEM media for overnight. Thereafter, cells were starved with serum-free DMEM for 12 h. Cells were then incubated with fresh serum-free DMEM containing LPA. After 12 h of incubation, conditioned media (CM) were collected, centrifuged to remove the cell debris if any, and was used to treat RAW264.7 cells.
Treatment of conditioned media
To induce osteoclastogenesis, RAW264.7 cells were cultured in differentiation media containing 30 ng/ml RANKL for 2.5 days. After that, cells were incubated with fresh Alpha-MEM (without RANKL) containing 50% Conditioned Medium (CM) for another 2.5 days. Serum-free DMEM (50%) was used as vehicle treatment. At day 5, cells were collected for RNA extraction or were stained for tartrate resistant acid phosphatase (TRAP assay) to observe osteoclast formation.
Cell viability assay
RAW264.7 cells were cultured in differentiation media for 2.5 days and treated with CM for another 2.5 days. Thereafter, media were replaced with fresh media containing MTT solution at a final concentration of 0.5 mg/ml. After 2 h of incubation at 37℃, supernatant was carefully removed and the insoluble formazan crystal was dissolved in 200 μl of dimethyl sulfoxide (DMSO). Optical density (OD) of the dissolved crystals was measured at 570 nm.
TRAP staining
At day 5, cells were fixed and stained for TRAP using the Acid Phosphatase Leukocyte kit (Sigma-Aldrich). The TRAP (+) multinucleated cells that contained three or more nuclei were counted as osteoclasts using Ziess AxioCam digital microscope.
Reverse transcription-quantitative polymerase chain reaction (RT-PCR)
Total RNA (RNAs) was extracted from cultured cells using Trizol reagent (Invitrogen, Carlsbad, CA). After that, 2 μg of RNAs was reverse-transcribed into cDNA using SuperScript II Reverse Transcriptase (Invitrogen). One μl of cDNA was diluted into 20 μl of qPCR reaction mixture containing SYBR Green (2xGreen star qPCR MasterMix, Bioneer, Korea). qPCR was performed on Rotor-Gene Q (Qiagen, Hilden, Germany) with cycling conditions as follow: initial denaturation at 95℃ for 10 min, and then 40 cycles of denaturation at 95℃ for 20 s, annealing at 60℃ for 20 s and extension at 72℃ for 25 s. Relative mRNA expression levels of the selected genes were analyzed using ΔΔCT method and were normalized to GAPDH. The sequences of qPCR primers are listed in Table 1.
Protein isolation and western blot analysis
Cells cultured in multiple well-plate were washed with PBS and lysed with cold RIPA buffer (20 mM Tris-HCl, pH 7.5, 200 mM NaCl, 1% Triton X-100, 1 mM dithiothreitol) containing protease inhibitor cocktail (Roche, Mannheim, Germany) and phosphatase inhibitors. Protein concentrations were determined using micro BCA protein assay kit (Thermo Scientific, Massachusetts, USA) following the manufacture's protocol. Total protein was separated on 9% SDS-polyacryramide gel electrophoresis and transferred to PVDF membrane (Millipore, Billerica, MA). The membrane was then blocked with 5% skimmed milk and incubated with primary antibodies as follow: polyclonal anti-rabbit p-PKC (1:1000 dilution in 1% BSA) and monoclonal anti-mouse β-actin (1:1000 dilution in 1% BSA) for 2 h at room temperature. After washing, membrane was incubated with horseradish peroxidase-conjugated anti-rabbit IgG secondary antibody (1:2000 dilution in 5% skim milk) or anti-mouse IgG secondary antibody (1:5000 dilution in 5% skim milk). Protein bands were visualized using Luminata Forte western HRP substrate (Millipore) and signals were detected and quantified using Fusion-Fxsystem (Vilber Lourmat, Eberhardzell, Germany).
ELISA
After 12 h of LPA stimulation, supernatant from both MDA-MB-231 and MDA-MB-468 cells were collected. Human IL-8 and IL-11 ELISA kit (R&D systems, Minneapolis, MN) were utilized for a quantitative measurement of cytokines according to the manufacturer's recommendations.
Statistical analysis
All the statistical data were analyzed by Graphpad Prism 5.0 (San Diego, CA) and evaluated using two-tailed Student's t-test. All data was presented as mean±SEM of at least three independent experiments. Value of p<0.05 was considered to indicate a statistically significant difference.
RESULTS
LPA induced expression of IL-8 and IL-11 in breast cancer cells
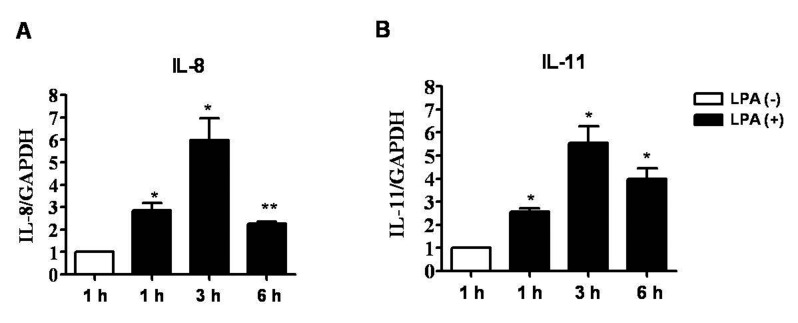
IL-8 and IL-11 have important roles in breast cancer progression and bone metastasis mechanism. To determine whether LPA stimulates expression of IL-8 and IL-11 in MDA-MB-231 breast cancer cells, serum-starved cells were treated with 10 μM of LPA and the expression of IL-8 and IL-11 mRNA were examined after 1 h, 3 h and 6 h of LPA treatment. A dose of 10 μM of LPA has been shown to induce no effect on cell cycle of the MDA-MB-231 cells, ruling out any possibility of proliferative or cytotoxic effect [21]. The result indicates that treatment with LPA caused significant increases in IL-8 and IL-11 expression levels in MDA-MB-231 cells with maximal increments of about 5–6 folds of untreated control (Figs. 1A and B).
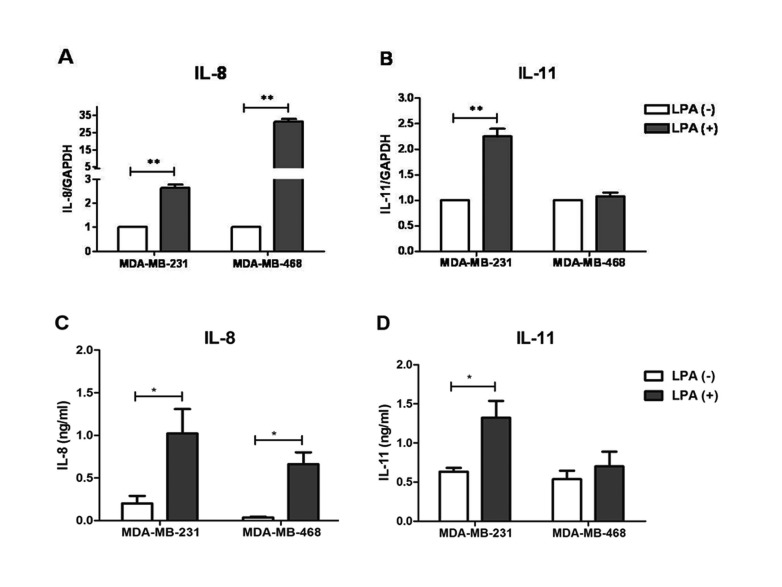
LPA has been known to act on breast cancer cells via it receptors, LPARs. Therefore, identification of specific LPA receptors responsible for induction of each osteolytic factor was required. It has been known that MDA-MB-231 and MDA-MB-468 are both triple-negative breast cancer cells. These cell types do not express three specific receptors: estrogen receptor (ER), human epidermal growth factor receptor 2 (Her2) and progesterone receptor (PR). These two cell lines differ from each other in expression of specific LPA receptors i.e; MDA-MB-468 cells predominantly express LPAR2 [1422] while MDA-MB-231 mainly expresses LPAR1 and LPAR2 [2324]. MDA-MB-231 is known as highly aggressive and invasive, poorly differentiated breast cancer cells. Compared to MDA-MB-231 cells, MDA-MB-468 cells are less aggressive and invasive (Table 2) [252627]. Both MDA-MB-231 and MDA-MB-468 were treated with LPA for 3 h and the mRNA expression level of IL-8 and IL-11 was analyzed by RT-PCR. We observed that treatment of LPA significantly increased the mRNA expression level of IL-8 in both the cell lines (Fig. 2A). LPA induced significant mRNA expression of IL-11 in MDA-MB-231 cells, but not in MDA-MB-468 cells (Fig. 2B). We also quantified the amount of IL-8 and IL-11 secreted from LPA stimulated MDA-MB-231 and MDA-MB-468 after 12 h of treatment (Figs. 2C and D). Similar to mRNA expression results, a significant increase of IL-8 was observed in the media of both the human breast cancer cell lines. However, amount of secreted IL-11 was only significant in MDA-MB-231 cells. The data shows that MDA-MB-231 cells expressing dominantly both LPAR1 and LPAR2, has induction of IL-8 and IL-11 after LPA stimulation, while, MDA-MB-468 cells expressing dominantly LPAR2, has induction of IL-8 only.
Analysis of signaling pathways involved in regulation of expression of IL-8 and IL-11 after LPA stimulation
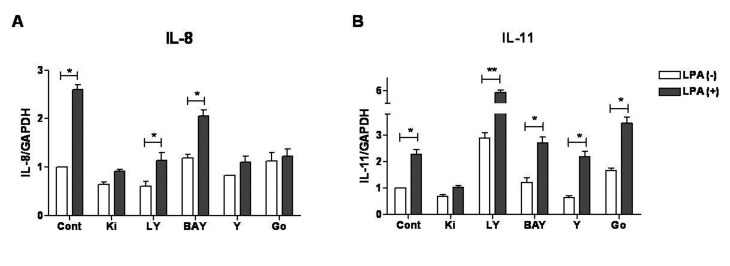
In addition to LPA receptors, we also hypothesized that specific signaling pathways might be involved in regulating expression of IL-8 and IL-11 in LPA-treated breast cancer cells. It has been reported that LPA induces IL-8 expression through signaling mechanisms like PI3K/Akt, ROCK, ERK, p38/MAPK, NFkB and JNK-dependent pathways in different cell types [28293031]. Similarly, IL-11 expression was found to be dependent on the activation of ERK, PKC, p38/MAPK and Smad signaling pathways [3233]. Therefore, involvement of these signaling pathways in LPA-induced IL-8 and IL-11 expression in MDA-MB-231 cells was examined. For this, inhibitor to specific pathways were used including ERK inhibitor PD98059 (10 μM), PI3K inhibitor LY294002 (10 μM), p38/MAPK inhibitor SB203580 (10 μM), JNK inhibitor SP600125 (2 μM), NFkB inhibitor BAY-11-7082 (5 μM), ROCK inhibitor Y27632 (5 μM) and PKC inhibitor Go6976 (1 μM). Moreover, a specific inhibitor for LPA receptors (Ki16425, 5 μM) was used to confirm the involvement of LPARs in induction of cytokines.
The data revealed that expression of both the cytokines was inhibited by LPARs inhibitor, Ki16425. Results also indicated that LPA-stimulated expression of both cytokines was not inhibited by ERK, p38/MAPK or JNK inhibitor (data not shown), confirming their non-involvement in LPA stimulated expression of both the cytokines. However, induction of IL-8 expression was strongly inhibited by ROCK and PKC inhibitors, also partially blocked by PI3K and NFkB inhibitors. But, none of these inhibitors affected the LPA-stimulated IL-11 expression (Fig. 3).
LPA induced IL-8 and IL-11 expressions were regulated by specific PKC subunits
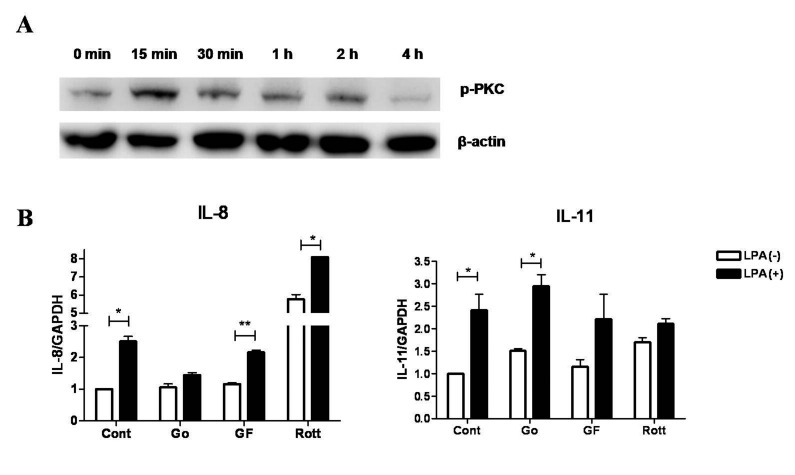
PKC is one of the main downstream signaling molecules for LPA receptors [18]. We observed the regulation of IL-8 induction by LPA via PKC pathway. Moreover, PKC has been reported in regulation of IL-11 in osteoblasts [34]. Hence, we examined activation of PKC after stimulation by LPA in MDA-MB-231 breast cancer cells. Cancer cells were treated with LPA for several time points from 15 min to 4 h. Phosphorylation of PKC was detected by western blotting. Fig. 4 shows that LPA increased level of p-PKC from 15 min to 2 h with maximal increment at 15 min and thereafter returning to normal level at 4 h after treatment.
Next, we aimed to clarify the involvement of PKCs subtypes in regulation of IL-8 and IL-11 expression after LPA stimulation. For this, different PKC inhibitors were used, including Go6976 (100 nM, selective inhibitor for PKCα, β1 and μ inhibitor), GF109203X (1 μM, selective inhibitor for PKCα, βI, βII, γ, δ and ε) and rottlerin (10 μM, selective PKCδ inhibitor) [35]. As shown in Fig. 4, IL-8 induction was strongly inhibited by Go6976 (similar result shown in Fig. 3), but not by GF109203X or rottlerin. However, IL-11 induction was inhibited by GF109203X and rottlerin. This result suggests that different PKC subtypes regulate expression of IL-8 and IL-11 in LPA-stimulated breast cancer cells. Namely, PKCμ is responsible for regulation of IL-8 because only PKCμ is inhibited by Go6976 but not by GF109203X or rottlerin, and PKCδ is responsible for IL-11 expression as it is inhibited by both GF109203X and rottlerin.
CM from LPA-stimulated breast cancer cells induced osteoclastogenesis
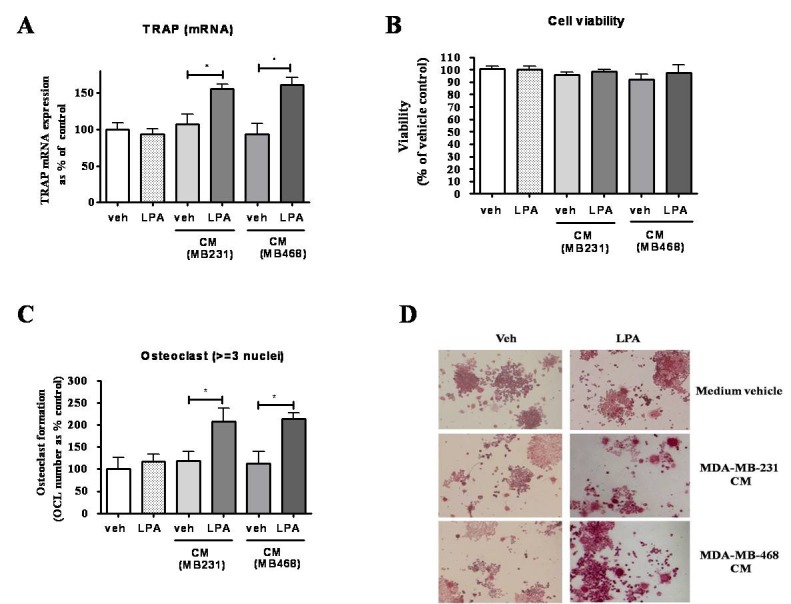
Upregulation of LPA signaling has been shown to enhance the formation of bone metastasis in mouse xenograft model [36]. Hence, we tried to inspect whether LPA-induced secretory factors from breast cancer cells can affect osteoclastogenesis. CM from breast cancer cells treated with or without LPA was collected and added to culture of RANKL-stimulated RAW264.7 cells. Results shows that LPA-treated breast cancer cells CM (MDA-MB-231 and MDA-MB-468) induced expression of differentiation marker, TRAP (about 1.5 folds), and incited formation of significant number of mature osteoclasts (about 2 folds), compared to vehicle CM. CM did not confer any effect on cell viability of RANKL-differentiated RAW264.7 cells in comparison to vehicle CM. To order to exclude the possibility that LPA existing in CM, due to treatment, might affected osteoclastogenesis, we directly added LPA into culture of RAW264.7 cells. The result shows that LPA did not affect osteoclastogenesis, compared to vehicle control (Figs. 5A–D).
DISCUSSION
LPA, a growth factor-like phospholipid has diversity of effects on cell physiological activities in many cell types. Role of LPA in cancer progression has been elucidated in numerous studies. Recently, studies on the involvement of LPA on cancer metastasis such as bone invasion has become an emerging research area [20].
Our study analyzed the possible role of LPA in breast cancer cells-mediated osteoclastogenesis via upregulation of osteoclastogenic cytokines, IL-8 and IL-11. Treatment of LPA to MDA-MB-231 cells induced the expression of IL-8 and IL-11 (Figs. 1 and 2). LPA-induced IL-8 expression and secretion was reported in several cell types including breast cancer cells [282930]. Moreover, breast cancer cells stimulated with TGFβ displays increased expression of IL-8 and IL-11 [3237]. Our result revealed that LPA also induces IL-11 expression in MDA-MB-231 cells. Similar to previous study of Mu et al, LPA stimulation increased the mRNA expression of IL-8 (maximally, about 6 folds) after 3 h of treatment [38]. Moreover, our results also demonstrates that LPA enhanced mRNA expression of IL-11 up to 5-6 folds at the same time point. ELISA results also confirmed the induction of IL-8 and IL-11 in protein after LPA stimulation (Figs. 2C and D).
Since, LPA induced the expression level of IL-8 and IL-11 in MDA-MB-231 breast cancer cells, association of LPA-stimulated expression of these cytokines with LPARs and downstream signaling pathways of LPARs were investigated. In breast cancer, LPARs are expressed in many cancer cell lines, but at various levels. Till now, there are only few studies addressed the involvement of LPARs and cytokine expression. Our study suggests that LPAR1 might be responsible for IL-11 and LPAR2 for IL-8 induction in stimulated Triple-Negative Breast Cancer (TNBC) cells (Fig. 2). LPAR2 is highly expressed in less aggressive breast cancer cells (MDA-MB-468) than in more aggressive cells (MDA-MB-231 cells) [1422]. This might explain for a strong induction of IL-8 in MDA-MB-468 (about 30 folds) after LPA treatment. Moreover, our result is consistent with previous study of Hartman et al, indicating that regulation of IL-8 is mediated by LPAR2 in TNBC cells [30]. The other evidence for involvement of LPARs in LPA-stimulated cytokines expression is that treatment with LPA receptors inhibitor (Ki16425) reduced the induction of IL-8 and IL-11 in breast cancer cells (Fig. 3). Thus, it may be concluded that specific LPA receptors might be involved in regulating the induction of IL-8 and IL-11 expression in TNBC cells. Recently, a number of naturally occurring or synthetic analogue of LPA has been recognized by various studies [3940]. These LPA analogues promises to be a selective type of LPA receptor ligand (agonist or antagonist) providing an opportunity to probe complex overlapping LPA signaling pathways. Further studies might utilize them for bifurcating the complex LPA receptors signaling mechanism in induction of cytokines for bone metastasis by breast cancer cells.
LPARs have been known as G protein-coupled receptors. Activation of LPARs leads to activation of three different G alpha subunits Gi/o, Gq, G12/13, in turn, it triggers activation of many cellular signaling such as PKC, Ras/ERK, PI3K/Akt, RhoA/ROCK and NFkB [15]. Our study indicates that IL-8 induction was mostly dependent on ROCK, PKC, and to a lesser extent on PI3K and NFkB signaling pathways. Previous study have reported that LPA-regulated IL-8 secretion in Sum159, a TNBC cell line, was completely inhibited by the treatment of NFkB inhibitor, BAY-11-7082 (10 μM) [30]. The difference between our study and previous study may be due to the difference in dose of inhibitor used for the treatment. In our case, we have used a lower dose of NFkB inhibitor (5 μM) while other study used a higher dose of 10 μM. Moreover, the difference in the cell type utilized in the two studies might be a factor. However, the signaling pathways responsible for IL-8 induction were not involved in the regulation of IL-11.
PKCs belong to a serine/threonine kinase family which includes three classes of isoenzymes: conventional isoforms (α, β and γ) that are calcium-dependent and require diacylglycerol (DAG) for activation; novel isoforms (δ, ε, η, θ and μ) that are calcium-independent but requires DAG for activation; and atypical isoforms (ζ, ι, and λ) that are independent of both Ca2+ and DAG. It was observed that MDA-MB-231 breast cancer cells express high levels of PKC subtypes [41]. In our study, LPA treatment induced rapid activation of PKC as observed by the increase in the levels of p-(pan) PKC from 15 min to 2 h of treatment (Fig. 4A). The normalization of phosphorylation of PKC at 4 h of treatment of LPA implies it can only stimulate PKC signaling pathway at a very early time point and is for a short duration. Moreover, the result obtained shows that just PKCμ and no other PKC subtypes are involved the regulation of IL-8 expression in LPA-stimulated breast cancer cells (Fig. 4B). For the first time, this study indicated a possibility that LPA-induced IL-11 expression in breast cancer cells might involve the PKCδ subtype (Fig. 4B).
Breast cancer cells have been known to secret many factors including osteolytic factors. Among osteolytic factors, cytokines IL-8 and IL-11 are mostly implied in breast cancer for bone metastasis. Our results demonstrated that LPA-stimulated breast cancer cells CM increased the expression of osteoclast differentiation marker, TRAP, in macrophage cell line, as well an increase in the number of mature osteoclasts were also observed (Fig. 5). It appears that secreted products from LPA-stimulated breast cancer cells can promote osteoclastogenesis. Since, our RT-PCR data showed an enhanced mRNA expression of IL-8 and IL-11 after stimulation of breast cancer cells, role of IL-8 and Il-11 in increased osteoclastogensis can be assumed.
Metastasis is complex process in which breast cancer cells leave the original tumor site and migrate into other part of body via circulatory systems (bloodstream or lymphatic system). In the current study, LPA increased the expression levels of IL-8 in MDA-MB-231 and MDA-MB-468 cells and conditioned media from LPA-stimulated breast cancer cells MDA-MB-231 and MDA-MB-468 increased osteoclastogenesis, therefore, IL-8 might mediate osteoclastogensis in this context. In addition, as shown in Table 2, MDA-MB-231 cells are highly aggressive, metastatic cells and LPA-stimulated MDA-MB-231 cells expressed higher levels of IL-11, suggesting possible role of IL-11 in metastasis. LPA is a component of serum, mainly generated from platelets and breast cancer cells are also known to secret LPA. Hence, it can be presumed that elevated presence of LPA in tumor micro-environment can stimulate breast cancer cells via an autocrine pathway to help in metastasis process. However, more detailed studies will be required to delineate the exact mechanism of induction of cytokines by LPA stimulated breast cancers and their participatory role in metastasis. Better understanding of mechanism of LPA regulation of its products may support to finding the good treatment for breast cancer metastasis.


 XML Download
XML Download