

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cardiac conditioning is a therapeutic approach to induce an endogenous protective adaptive response in the heart for counteracting sustained ischemia-reperfusion injury [12]. In this approach, heart or other remote organ is subjected to short period of ischemia and reperfusion (conditioning stimulus) either before or after sustained ischemia. Based on the period of delivery of the conditioning stimulus (before, during or after sustained ischemia), and site of delivery (to the target organ or remote organ), the treatment approach is classified into ischemic preconditioning [3], ischemic perconditioning, ischemic post-conditioning [4], remote preconditioning [5] and remote postconditioning [6]. Pharmacological preconditioning is a treatment approach wherein the administration of various pharmacological agents elicits preconditioning like cardioprotective effects [7].
The aging process induces a plenty of changes in the composition of body by altering the energy balance and may increase the susceptibility of individuals to the cardiovascular diseases [89]. Despite the advances in prophylactic and preventive therapies, cardiovascular disorders remain one of the main causes of deaths in the elderly cohorts [10]. There have been abundant preclinical reports indicating that aging increases the vulnerability of the heart to ischemia-reperfusion injury [1112]. Furthermore, various researchers have demonstrated that aging reduces the development of adaptive cardioprotective response on being subjected to conditioning stimuli, including hypoxic or ischemic preconditioning [131415], ischemic postconditioning [16] and remote ischemic preconditioning [17]. The present review describes aging as one of the major confounding factors that may attenuate conditioning-induced cardioprotection along with the possible mechanisms.
EVIDENCES
Aging exaggerates ischemia-reperfusion injury
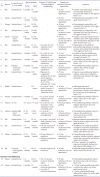
Aging is one of the major prognostic determinants of myocardial infarction and renders the aged heart more susceptible to coronary artery disease [181920]. Lesnefsky et al. reported that aging increases the susceptibility of the isolated heart to ischemia-reperfusion injury. The authors reported that aging significantly reduces the recovery of left ventricular developed pressure (LVDP) following ischemic injury in aged rats (24 months). Furthermore, tissue damage in terms of lactate dehydrogenase (LDH), creatine kinase (CK) release and total protein release was much higher in the aged rat hearts [21]. Similarly, Tani and coworkers reported a gradual decline in ischemic tolerance with increasing age in isolated rat hearts. Sustained ischemia and thereafter reperfusion led to greater impairment in functional contractility, enhanced LDH and CK release in the aged rat hearts (50–100 weeks) in comparison to the young hearts (12 weeks) [22].
Scientists have also reported that there is enhancement of ischemia reperfusion-induced myocardial injury due to ageing in mice. Azhar et al. demonstrated that left anterior coronary artery ligation followed by reperfusion led to greater myocardial necrosis, DNA fragmentation, and mitochondrial disruption in the old mice hearts (22–24 months) in comparison to the young mice hearts (6–8 months) [23]. Other studies have also reported that senescence substantially reduces the ischemic tolerance in the mice hearts (18–28 months) [1124]. Furthermore, it also shown that the deleterious effect of co-morbidity on ischemia reperfusion injury is significantly increased in aged animals. Whittington et al. reported that the aged diabetic Goto-Kakizaki rats (18 months) are more prone to ischemic injury in comparison to the young diabetic rat hearts (3 months) [15].
Apart from the studies on animals, Mariani et al. reported that aged human myocardium also loses the capacity to recover following hypoxic injury. The authors reported the lesser degree of recovery (in terms of development of force) in the atrial pectinate trabeculae (dissected from the right atrial appendages of the patients, 34–89 years) isolated from the old cohorts (around 89 years) in comparison to the young cohorts (around 34 years) in response to hypoxic injury (Table 1) [25].
Aging attenuates the protective effects of ischemic preconditioning
In animals: Apart from studies showing per se deleterious effects of aging on ischemia reperfusion injury, studies have also shown that aging attenuates the cardioprotective effects of ischemic preconditioning [14152627]. Fenton and co-workers reported that two episodes of ischemic preconditioning (5 min ischemia-5 min reperfusion) prior to ischemic insult (45 min ischemia-120 min reperfusion) significantly reduced the development of necrosis and remarkably improved the post-ischemic functional recovery in the isolated young adult (3–4 months) rat hearts (Fischer 344). However, ischemic preconditioning stimulus failed to attenuate the development of necrosis and impaired the contractile function in the aged hearts (21–22 months) suggesting that aging impairs the ability of preconditioning stimulus to reduce necrosis and contractile dysfunction in the rat hearts [26]. Schulman et al. reported that preconditioning stimulus led to reduction of infarct size in the young rat hearts (3 months). However, the senescent hearts (18–20 months) were refractory to ischemic and pharmacological preconditioning with 2-chloro-N(6)-cyclopentyladenosine (adenosine A1 receptor agonist), 1,2-dioctanoyl-sn-glycerol (the protein kinase C agonist) and diazoxide (mitochondrial KATP-channel opener). This indicates that aging induces alterations within ischemic preconditioning signaling cascade to reduce its ability to produce cardioprotection [27]. Tani et al. reported that hypoxic preconditioning stimulus improves post-ischemic recovery and reduces CK release selectively in the young rat hearts, but not in the senescent hearts [28]. Besides, Boengler and co-workers also reported that ischemic preconditioning (10 min ischemia and 10 min reperfusion) significantly reduced ischemia-reperfusion-induced myocardial infarct size in the young hearts (<3 months), without producing protective effects in the aged mice hearts (>13 months) [29].
McCully et al. reported the novel protective protocol termed as “adenosine-enhanced ischemic preconditioning” in which adenosine was given along with ischemic preconditioning to enhance the cardioprotection [30]. These authors reported that the combination of adenosine with ischemic preconditioning significantly produced cardioprotection in senescent rabbit hearts and also produced functional recovery in these hearts. Based on this, it may tentatively proposed that decrease in adenosine levels due to aging may possibly contribute in attenuating the cardioprotective effects of ischemic preconditioning. Therefore, addition of adenosine in ischemic preconditioning protocol produced cardioprotection in aged hearts [31].
Cells of animals: The studies performed on cardiomyocytes have also revealed that cardioprotective effects of ischemic preconditioning are lost due to aging. Brien and Howlett reported that ischemic preconditioning of ventricular myocytes isolated from young adult Fischer 344 rats significantly abolished postischemic contractile dysfunction, diastolic contracture and markedly improved the survival of myocytes. Moreover, ischemic preconditioning also increased the amplitude of Ca2+ transients reperfusion and diastolic Ca2+ during reperfusion. Although, ischemic preconditioning increased the amplitude of Ca2+ transients, improved contractile function, reduced the marked rise in diastolic Ca2+ in the aged myocytes during reperfusion, yet it failed to improve the survival of these aged myocytes. This indicates that increased availability of intracellular Ca2+ during preconditioning possibly improves the contractile dysfunction. However, the decrease in survival of the aged myocytes can be possibly due to age-dependent changes in the structure and functioning of the isolated myocytes [14].
Preconditioning stimulus tends to reduce platelet adhesion and aggregation in models of myocardial infarction and recurrent thrombosis. The anti-platelet effect of ischemic preconditioning is possibly due to enhanced discharge of adenosine and subsequent activation of adenosine A2 receptors on the platelet surface [32]. Przyklenk and Whittaker demonstrated that exogenous administration of CGS 21680 (A2 receptor agonist) in young adult rabbits led to reduction of platelet aggregation in an in vitro model of platelet aggregation. However, CGS 21680 administration failed to reduce platelet aggregation in the old cohort indicating age-dependent reduction in responsivity of the platelets to adenosine preconditioning [33].
Humans and human tissues: Apart from this, various researchers have reported that ischemic preconditioning does not produce cardioprotective effects in the senescent human myocardium [343536]. The existence of angina before acute myocardial infarction serves as preconditioning stimulus (physiological preconditioning) and exerts cardioprotective effect in the adult patients. However, the protection afforded by antecedent angina is lost in the senescent myocardium indicating absence of preconditioning like protective effects in the old cohorts [34]. Furthermore, Wu et al. reported that ischemic preconditioning stimulus protects the heart against ischemia-reperfusion injury in adult patients undergoing coronary artery bypass grafting in terms of reduced cardiac troponin I release and improvement in ejection fraction and cardiac index. However, preconditioning stimulus failed to reduce cardiac troponin I release, improve ejection fraction and cardiac index in the aged patients undergoing coronary artery bypass grafting. This again indicates that the protective effect of preconditioning stimuli diminishes with age [35]. The loss of protective effects of ischemic preconditioning with increasing age is further supported by a study of showing that ischemic preconditioning did not improve functional recovery of right atrial trabeculae of old patients (more than 70 years age) in comparison to adult patients (less than 55 years) [36]. Another study reports that aging significantly reduces ischemic preconditioning-induced protective effects on endothelial cells. The authors reported that ischemic preconditioning selectively preserved flow-mediated dilation in the brachial artery in young men (20–25 years), but not in the elderly patients (68–77 years) suggesting that aging reduces the ability of ischemic preconditioning to protect against ischemia-reperfusion injury associated endothelial dysfunction [37]. On the contrary, a study of Loubani et al. on right atrial tissues isolated from hearts undergoing elective heart surgery indicated that aging does not influence the protective effects of ischemic preconditioning and ischemic preconditioning was shown to protect atrial tissues of all age groups against ischemia-reperfusion injury (Table 1) [38].
Aging abolishes remote ischemic preconditioning-induced cardioprotection
It has also been documented that aging is a confounding factor in reducing the effectiveness of remote ischemic preconditioning-induced adaptive response against ischemia-reperfusion injury in rodents [17]. Behmenburg et al. reported that four cycles of remote ischemic preconditioning (5 min hind limb ischemia-5 min reperfusion) prior to sustained ischemia (25 min) and 120 min reperfusion led to significant reduction in the infarct size in young adult rats (2–3 months). However, remote ischemic preconditioning-dependent reduction in infarct size was absent in senescent heart (22–24 months). This indicates that the efficacy of remote ischemic preconditioning to reduce infarct size reduces with age [17]. Moreover, Hausenloy et al. reported that remote ischemic preconditioning failed to improve clinical outcomes in patients (n=1612) undergoing coronary artery bypass graft surgery. The authors reported that the incidences of myocardial infarction, coronary revascularization, stroke and death in older adult patients (76.3±7.0 years) were not reduced even after remote preconditioning. Furthermore, remote ischemic preconditioning did not reduce troponin T levels, inotropic score, acute kidney injury and total stay period in the intensive care unit suggesting that the efficacy of remote ischemic preconditioning may not be observed in older patients (Table 1) [39].
Negative influence of aging on pharmacological preconditioning and postconditioning
As per reports of ischemic and remote preconditioning, the effects of pharmacological preconditioning and postconditioning are also abolished with increasing age. Sniecinski and Liu reported that sevoflurane preconditioning significantly reduced CK release, infarct size and improved left ventricular developed pressure following 25 min of ischemia and 60 min of reperfusion in young rats, but not in the aged rats [40]. Peart and Gross demonstrated that acute infusion of morphine significantly improved left ventricular developed pressure and end diastolic pressure in the young (10–14 months), but not in the senescent mouse hearts (24–26 months) [41]. Nguyen et al. reported that pharmacological preconditioning with isoflurane, an anesthetic, abolished coronary artery occlusion-induced increase in myocardial infarct size in the young rats 0 (3–5 months) in comparison to old rats (20–24 months) [42]. Similarly, Li et al. reported that sevoflurane postconditioning followed by 30 min ischemia and 120 min reperfusion did not reduce infarct size in senescent rats (20–24 months) in comparison to the young rats (3–4 months) [43]. On the contrary, Jiang and co-workers reported that sevoflurane postconditioning significantly improved coronary flow, functional recovery, reduced mitochondrial lesion severity and apoptosis in the aged rat hearts (Table 1) [44].
Aging as a confounder in ischemic postconditioning
Przyklenk et al. reported that repeated cycles of ischemic postconditioning reduce the infarct size in isolated adult mouse hearts (3–4 months), while it failed to reduce the infarct size in the aged mouse hearts (20–24 months). This suggests that aging reduces the effectiveness of ischemic postconditioning in providing protection against myocardial necrosis [16]. Boengler et al. reported that three cycles of ischemic postconditioning reduced the infarct size in the young mice (3 months), without any significant effect in aged mice (>13 months). However, augmented ischemic postconditioning i.e., five cycles (instead of three cycles) of ischemic postconditioning reduced infarct size in the aged mice hearts (>13 months). This indicates that an increase in the number of cycles of ischemic postconditioning stimulus may overcome the confounding effects of aging (Table 1) [45].
MECHANISMS RESPONSIBLE FOR AN INCREASE IN ISCHEMIC INJURY DUE TO AGING
Estrogen deficiency
Estrogen status of a female rat determines the susceptibility of the heart to ischemia-reperfusion injury [4647]. Aging leads to a significant decline in the estrogen levels in the females and increases the susceptibility of the heart to ischemia-reperfusion injury [4849]. Xu et al. reported that chronic in vivo administration of estrogen in aged ovariectomized rats (12–14 months) markedly improved functional recovery after 25 min ischemia-40 min reperfusion [48]. In corroboration with the above study, Novotny et al. described that selective estrogen receptor-α activation in aged female rats partly reduces infarct size [50]. A previous study has also indicated that administration of 17β-estradiol significantly reduces myocardial infarct size in canine hearts [51]. Furthermore, a study has indicated that administration of 17β-estradiol in ovariectomized female rats significantly increased the number of viable myocytes and reduced mitochondrial and ultrastructural damage to the myocytes isolated from the hearts [46]. Apart from these, clinical trials have shown that estrogen levels decline with age [52] and postmenopausal hormone replacement therapy tends to reduce mortality after myocardial infarction [53]. Researchers have shown that estradiol prevents the ischemia-induced release of cytochrome c from mitochondria, prevents caspase activation and apoptosis [54]. Furthermore, Kam et al. reported that estrogen confers cardioprotective effects possibly via suppressing the β1-adrenoreceptor expression [55]. Based on these studies, it may proposed that a decrease in estrogen levels with aging may contribute in augmenting ischemic injury by promoting myocardial apoptosis and increasing the expression of excitatory β1-adrenoreceptors on heart.
Reduction in STAT3 phosphorylation
Aging markedly inhibits the activation of signal transducer and activator of transcription (STAT) signaling pathway and increases the susceptibility to ischemia-reperfusion injury in the heart. STAT3 are transcription factors that play pivotal role in cytoprotective signaling and are activated in the myocardium exposed to ischemia-reperfusion injury [565758]. Hilfiker-Kleiner revealed that STAT3 deficient mice exhibit increased susceptibility to myocardial ischemia-reperfusion injury [59]. Furthermore, Oshima and co-workers reported that transgenic mice constitutively expressing active STAT3 gene had reduced ischemia-reperfusion injury. Meanwhile, formation of reactive oxygen species (ROS) was significantly decreased in transgenic mice and the expression of metallothionein 1 and metallothionein 2 (ROS scavengers) was up-regulated in the transgenic hearts. However, homozygous deletion of metallothionein1 and metallothionein2 markedly reduced the cardioprotective effect of STAT3 against ischemia-reperfusion injury. This indicates that activation of STAT3 protects the heart against ischemia-reperfusion injury probably via metallothionein 1 and metallothionein 2 [60]. Therefore, it may be hypothesized that aging may decrease the phosphorylation of STAT3, which may in turn decrease the levels of metallothionein 1 and metallothionein 2 and increase the levels of reactive oxygen species to augment ischemia-reperfusion induced myocardial injury.
Impaired mitochondrial function, autophagy and increased ROS formation
Autophagy has a critical role in maintaining cardiac structure and function [61]. Mitochondria regulate biological signals for autophagy and maintain a healthy pool of mitochondria by removing damaged mitochondria through autophagy [6263]. Dutta and co-workers reported that aging results in progressive decline in the cardiomyocyte mitochondrial function and reduces macroautophagy, which leads to accumulation of damaged mitochondria within the cardiomyocytes. This is accompanied by reduced ATP production and increased formation of ROS [64]. In corroboration with the above report, Fernandez-Sanz et al. reported that aged hearts display greater ischemia-reperfusion injury and there is a rapid decline in the mitochondrial membrane potential in comparison to the young hearts. Furthermore, the authors reported a reduction in mitochondrial Ca2+ uptake and increased cytosolic Ca2+ overload in the aged cells [65]. This contention is supported by a previous study conducted by Duicu et al. indicating that aging significantly reduces mitochondrial membrane potential and increases ROS production. Furthermore, the authors reported that aging markedly increases the mitochondrial sensitivity to Ca2+-induced mPTP opening [66]. Accordingly, it may be proposed that aging may decrease the process of autophagy, which may lead to accumulation of damaged mitochondria. It may produce mitochondrial dysfunction, which may be manifested in the form of an increase in production of reactive oxygen species, induction of apoptosis and increase in cytosolic calcium levels to augment myocardial injury.
Reduced glycogen synthase kinase-3β phosphorylation (GSK-3β) and protein kinase C (PKC) signaling
Glycogen synthase kinase-3 is a key regulatory enzyme for glucose metabolism which is inactivated on phosphorylation and exists as an active protein kinase in the basal state [67]. An increase in GSK-3β phosphorylation and subsequent inhibition of GSK-3β activity protects the young myocardium against sustained ischemia-reperfusion injury [68]. However, aging tends to reduce the ischemic tolerance possibly due to decreased GSK-3β phosphorylation and decrease in the levels of p70s6K (pro-survival signaling kinase, which inhibits GSK-3β) in aged murine myocardium [69]. The above contention is further supported by the fact that administration of GSK-inhibitor in the aged rats failed to induce cardioprotective effects in comparison to the younger rats [70]. Furthermore, authors reported that senescence possibly reduces PKC signaling to decrease the ischemic tolerance [5071]. It is possible that there may be a decrease in levels of pro-survival kinases, which may decrease GSK-3β phosphorylation and increase GSK-3β activity to augment myocardial injury.
Reduction in thioredoxin activity
Thioredoxin is an antioxidant redox protein that is an important antiapoptotic molecule and tends to reduce myocardial ischemia-reperfusion injury [72]. Aging significantly decreases thioredoxin activity and increases the susceptibility to myocardial ischemia-reperfusion injury [73]. Furthermore, thioredoxin expression and thioredoxin nitration (posttranslational modification that inhibits thioredoxin activity) was markedly increased in the aging heart [73]. Therefore, it is hypothesized that reduction in thioredoxin activity potentially contributes in increasing the risk to ischemia-reperfusion injury.
Based on all these proposed mechanisms, it may be proposed that aging may promote the production of free radicals and induce apoptotic cell death due to mitochondrial dysfunction, decrease in estrogen, decrease in STAT3 phosphorylation, decrease in antioxidants including metallothionein and thioredoxin to exaggerate ischemia-reperfusion-induced myocardial injury.
MECHANISM RESPONSIBLE FOR REDUCED EFFECTIVENESS OF DIFFERENT CONDITIONING STIMULI
Ischemic preconditioning
Aging leads to progressive loss of mechanisms underlying ischemic preconditioning-induced cardioprotection.
Impairment in adenosine functioning: It has been well documented that preconditioning stimulus induces adenosine release, which subsequently activates myocardial adenosine receptors to produce cardioprotection [247475]. The catabolism of ATP releases adenosine into the interstitial compartment of the myocardium, which is then partitioned between the intracellular compartment and interstitial space of the heart via Na+-dependent and Na+-independent transporters [76]. Lorbar et al. revealed that aging reduces the uptake of adenosine into the cardiomyocytes from the interstitial compartment [77]. In corroboration to the above study, Dobson and Fenton also reported that aging increases interstitial concentration of adenosine in the heart. The authors also reported that an increase in the adenosine levels in interstitium exerts anti-adrenergic action and may reduce the responsiveness to adrenergic stimulation [78].
Apart from the decrease in uptake of adenosine into cardiomyocytes, there may be the reduction in the availability of mitochondrial AMP to produce adenosine [79]. The loss of cardiac conditioning response in the aged hearts may also be due to insensitivity of adenosine A1 and A3 receptors [69]; down-regulation of transcription and expression of adenosine A1, A2 and A3 receptors [1180] and reduced coupling between A1 receptors and G-proteins [81]. Indeed, it has been reported that A1 over-expression overcomes age related failure in signaling and restores adenosine mediated cardioprotection [82]. It is also documented that activation of adenosine receptors reduces free radical formation via opening of KATP channels [83]. Therefore, it is possible that the reduction in adenosine release or its impaired functioning may increase the generation of free radicals to attenuate the cardioprotective effects of ischemic preconditioning during aging.
Increase in oxidative stress: As discussed in previous sections, aging is associated with increased formation of free radicals in the myocardium [84]. Whittington et al. reported a significant decline in the expression of PGC-1α (Peroxisome proliferator-activated receptor-gamma coactivator) and catalase (anti-oxidant enzyme) in the aged diabetic rat hearts during ischemic preconditioning [15]. PGC-1α regulates the transcriptional activity of antioxidant genes in including catalase [85]. Thus, increased oxidative stress in the form of reactive oxygen species/reactive nitrogen species may contribute to aging dependent loss in responsiveness to the conditioning stimuli in the senescent diabetic rat hearts [15]. Lucas and Szweda reported that aging increases the concentration of 4-hydroxy-2-nonenal, HNE (major product of lipid peroxidation) in the cardiac tissue upon exposure to ischemia and reperfusion, which modifies proteins and mitochondrial respiration [86]. In concordance with the above study, Lesnefsky and co-workers revealed that aging increases the rate of oxidative phosphorylation and the activity of electron transport complex 3 in the cardiac mitochondria of aged rats indicating that aging increases oxidative stress [87]. Therefore, an increase in oxidative stress through multiple mechanisms may contribute in attenuating ischemic preconditioning-induced cardioprotection during aging.
Reduction in connexin 43 content in the myocardium: Connexin 43 is the major constituent protein of myocardial gap junctions, and it is implicated in mediating ischemic preconditioning-induced cardioprotection [2888]. Boengler and co-workers demonstrated that ischemic preconditioning significantly increases the mitochondrial connexin 43 content in the atrial and ventricular tissue homogenates of the young mouse hearts, but not in the aged mouse hearts [28]. Furthermore, Schwanke et al. reported that ischemic preconditioning did not induce cardioprotective effects in the cardiomyocytes isolated from connexin 43 deficient mice indicating that connexin 43 significantly contributes in inducing in vivo cardioprotective effects of ischemic preconditioning [8990]. In addition, Li et al. reported that besides maintaining intercellular communication through gap junctions, connexin 43 also maintain volume homeostasis [91] and are necessary for providing preconditioning-induced cardioprotection. Therefore, it may suggested that aging may impair the process of augmenting the connexin 43 levels in the myocardium, which may contribute in attenuating the cardioprotective effects of ischemic preconditioning.
Kinase signaling: The activation of pro-survival kinases in response to the conditioning stimuli is essential to provide cardioprotection and counteract sustained ischemia-reperfusion injury [92]. Studies have strongly indicated the enhancement of Akt dependent signaling cascade [9394] in eliciting adaptive conditioning-induced cardioprotective response against ischemia reperfusion injury. Whittington et al. revealed that ischemic preconditioning stimulus fails to up-regulate Akt phosphorylation in the aged diabetic rat hearts in comparison to the young rat hearts [15]. Furthermore, Ledvenyiova and co-workers reported that gender and age also influence the activation of Akt signaling pathway during ischemic preconditioning [95]. Tani and co-workers reported that ischemic preconditioning fails to translocate protein kinase C (PKC) isoform from the cytosolic/membrane to the perinuclear region [96]. In corroboration with the above study, Peart et al. reported that aging leads to dysfunction of cardiac PKC signaling [69]. This indicates that aging possibly results in reduced phosphorylation of pro-survival kinases such as Akt and PKC, which is manifested in the form of decreased cardioprotective effects of ischemic preconditioning.
Moreover, aging also markedly inhibits the activation of JAK-STAT (Janus kinase and two signal transducer and activator of transcription) signaling cascade. STAT 3 is involved in the transduction of stress signals during ischemia-reperfusion injury from the plasma membrane to the nucleus to induce transcriptional changes in the genes [97]. Hattori and co-workers demonstrated that ischemic preconditioning increases the phosphorylation of JAK2 and STAT3; increases the expression of bcl-2 (antiapoptotic gene) and down-regulates bax (pro-apoptotic gene). These changes were significantly abolished in the presence of tyrphostin AG490, a selective JAK2 inhibitor suggesting that the activation of JAK-STAT signaling pathway during ischemic preconditioning may prevent myocardial apoptosis during ischemia-reperfusion injury [98]. The above contention was further supported by the results showing the attenuated effects of ischemic preconditioning in STAT3-deficient cardiomyocytes [99]. However, experimental studies are required to confirm that there is decrease in STAT3 expression in aged hearts, which directly attenuates the cardioprotection offered during ischemic preconditioning.
Alterations in ionic balance: The cardiac action potential relies on the ionic gradients set up by ion channels, and cardiac ionic balance disturbance may increase the vulnerability to ischemia-reperfusion injury [22100]. Tani et al. documented that aged rats are more susceptible to ischemia-reperfusion injury possibly due to alterations in the myocardial ionic balance. Aging significantly increases the intracellular levels of Na+ and K+ in the myocardium, which potentially decreases the cardioprotective response to hypoxic preconditioning in the senescent heart [29].
Increase in caveolin: Caveolae are cholesterol and sphingolipid enriched invaginations of the plasma membrane. Caveolins are the structural proteins required for caveolae formation and these exist in three isoforms viz. caveolin-1, caveolin-2 and caveolin-3 [101]. Caveolae can sense extracellular stress such as ischemia, flow-induced mechanical stretch and activate multiple signaling pathways [102]. Ajmani and co-workers reported that ischemic preconditioning fails to provide cardioprotection in diabetic rat hearts possibly due to up-regulation of caveolin and subsequent, reduction in eNOS activity [103]. Similarly, another study has reported that ischemic preconditioning fails to provide cardioprotection in ovariectomized rats possibly due to up-regulation of caveolin and decline in eNOS activity [104]. Moreover, it is also documented that the levels of caveolin-1 and caveolin-3 are increased with aging in the cardiomyocytes [105]. Therefore, it may be hypothesized that aging may increase the caveolin content in heart, which may reduce NO signaling to attenuate the cardioprotective effects of ischemic preconditioning. However, experimental studies are required to support this hypothesis.
Increase in phosphatase activity: Ischemic preconditioning promotes phosphorylation of signal transduction proteins to induce cardioprotective effects. However, aging tends to reduce the phosphorylation of these critical proteins in the aged rat myocardium (21–22 month). Fenton et al. reported that inhibition of endogenous protein phosphatases using okadaic acid (serine/threonine phosphatase inhibitor) restores preconditioning-dependent cardioprotective effects in the aged hearts. Furthermore, the authors reported that protein phosphatase 2A activity is greater in the aged ventricular myocardium in comparison to the young hearts (3–4 month) emphasizing the fact that aging-dependent protein dephosphorylation is responsible for reduction in ischemic preconditioning-induced cardioprotective effects [106].
Ischemic postconditioning
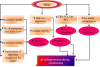
Kinases: As described in ischemic preconditioning section, ischemic postconditioning also significantly enhances STAT-3 phosphorylation in young mice to produce cardioprotection. However, these effects are abolished in the presence of AG-490 (JAK-2 Inhibitor) indicating that JAK phosphorylates and activates STAT-3 to induce cardioprotection. Moreover, ischemic postconditioning does not elicit cardioprotection in STAT-3 deficient hearts again suggesting the critical role of STAT-3 in ischemic postconditioning. However, STAT-3 phosphorylation is remarkably reduced in the aged mouse hearts [45], which emphasizes that reduction in STAT3 with aging may contribute in attenuating cardioprotection offered by ischemic postconditioning. Moreover, it has been shown that ischemic postconditioning stimulus fails to produce cardioprotection in old cohorts possibly due to decrease in ERK (a type of MAP kinase) and increase in MKP-1 (Mitogen-activated protein (MAP) kinase phosphatase-1) expression. MKP-1 is a protein phosphatases that deactivate mitogen-activated protein kinases by dephosphorylating both phospho-threonine and phospho-tyrosine residues. However, administration of sodium orthovanadate (non-specific MKP inhibitor) reduced MKP-1 expression, increased ERK expression and restored postconditioning-dependent cardioprotection (Fig. 1) [16]. Based on these, it may be proposed that decrease in JAK-STAT3 activity, increase in MAP kinase phosphatase and decrease in MAP kinase activities play an important role in attenuating cardioprotective effects of ischemic postconditioning in aged animals.
SUMMARY
Aging is one the major confounding factors that increases the susceptibility of the heart to ischemia-reperfusion injury. It is unlikely to assume that aging affects a single factor; rather multiple mechanisms may contribute in producing deleterious effects during aging. Aging tends to reduce the ischemic tolerance in the hearts due to multiple mechanisms including decline in estrogen levels in the females [4849]; decrease in autophagy, accumulation of damaged mitochondria, increase in reactive species formation, induction of apoptosis in the cardiomyocytes [64]; reduction in JAK-STAT-3 activity [59]; decrease in GSK-3β phosphorylation, increase in GSK-3β activity [70]; decrease in PKC signaling [5071] and decrease in antioxidants such as thioredoxin [73] and metallothionein [60].
Furthermore, it is manifested that aging decreases the development of adaptive response to various forms of conditioning stimuli (pre/post/remote conditioning). Adenosine release and subsequent activation of adenosine receptors during ischemic preconditioning exerts potent protective effects in the myocardium [2]. However, aging markedly impairs the ability of adenosine in preventing tissue damage during sustained ischemia through multiple mechanisms including impairment in the uptake and release of adenosine, decrease in the number adenosine transporter sites, down-regulation of the transcription of adenosine receptors in the myocardium and uncoupling of adenosine receptors with G-proteins [1177]. Decrease in adenosine functioning during ischemic preconditioning may trigger enhanced production of reactive oxygen species. Ischemic preconditioning stimulus also possesses the potential to activate antioxidant defense machinery to reduce the oxidative stress in the tissue [15]. However, aging tends to reduce the capability of the preconditioning stimuli to decrease the oxidative stress in the senescent hearts by decreasing the expression of antioxidant genes such as PGC-1α [15]. In addition, decrease in JAK-STAT-3 activity [98]; increase in MAP kinase phosphatase and decrease in MAP kinase expression; alteration in ionic balance (Na+ and K+) inside cardiomyocytes [29]; reduction in myocardial gap junction protein, connexin 43 content [28]; caveolin abundance and decrease in NO signaling [103104] and increases phosphatase activity leading to decrease in phosphorylation of cell signaling proteins [106] may contribute in attenuating ischemic preconditioning-induced cardioprotection in senescent hearts.
CONCLUSION
Aging markedly abolishes the potential of the preconditioning stimuli to induce cardioprotection due to impairment in adenosine functioning, increased oxidative stress, decrease in connexin 43 expression, caveolin abundance, reduced activation of kinases, STAT-3 and alteration in cardiac ionic balance. Aging also reduces the effectiveness of postconditioning stimuli possibly due to defective kinase signaling and reduced STAT-3 phosphorylation.
 XML Download
XML Download