

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Introduction
Emerging viral infections are a serious threat to the human population. During the Ebola virus disease (EVD) outbreak in West Africa in 2014–2016, thousands of people were infected and many of them died due to the viral infection [1]. EVD is generally considered as an endemic disease in Africa. However, its spread can be broadened through travel to all parts of the world. It is known to be transmitted from humans, bats, rodents, and chimpanzees to humans. Ebola virus belongs to the Filoviridae family [2]. Ebola virus has a negative sense RNA genome that encodes 7 different proteins and is surrounded by an envelope that contains the virally expressed glycoprotein (GP). There are five species of Ebola virus including, Zaire ebolavirus, Sudan ebolavirus, Tai Forest ebolavirus, Bundibugyo ebolavirus and Reston ebolavirus, all of which were named after the places where they were first discovered [3]. Ebola virus GP is associated with viral attachment to the host cell receptor prior to viral entry into the cells [45]. To date, neutralizing antibodies targeting Ebola virus GP have been reported to be somewhat effective at reducing the severity of Ebola virus infection and disease progression in animal models and humans. For example, when Ebola virus-infected monkeys were treated within 24 hours following Ebola virus challenge with ZMAb (composed of three monoclonal antibodies [MAbs] against Ebola virus GP), all survived from Ebola virus infection, while all monkeys without ZMAb treatment died within five days of viral infection [67] Furthermore, ZMapp (Mapp Biopharmaceutical) treatment is known to be effective at reducing mortality rates in patients infected with Ebola virus [8]. Despite the severity of Ebola virus infection, there are currently neither preventive vaccines nor therapeutic drugs readily available for use. Thus, development of vaccines or more effective drugs against Ebola viral infection is highly demanding.
MAbs have been broadly utilized as medicine, as well as diagnostic tools for virus infections. MAbs are generated by hybridoma technology where mice are injected with an immunogenic antigen and then the splenocytes are utilized to fuse with myeloma cells [9]. In this regard, antigens of high purity and with native protein folding are a pre-requisite for generating neutralizing antibodies that can target a native form of viral surface proteins. DNA vaccines are thought to be useful at generating the native from of viral antigens as they tend to express their proteins in cells in a manner similar to real viral infection. However, DNA vaccines are less effective at producing IgG-secreting hybridomas, possibly due to the presence of un-methylated CpG sequences in the DNA vaccines that stimulate polyclonal B cell activation [10]. In the Lee et al. (2017) study [10], use of protein boosting immunization was effective at reversing the preference for production of IgM-secreting hybridomas over IgG-secreting hybridomas. Moreover, multiple immunizations with GP after GP DNA vaccination might be useful at increasing the chance of generating antibodies with higher antigen binding affinity. In this context, it is highly likely that an appropriate application of GP DNA and protein vaccines may generate hybridoma clones that secrete IgG with higher binding affinity to the native form of the antigen.
In this study, we obtained five monoclonal IgG antibodies by an immunization scheme using Ebola virus GP DNA and protein vaccines. The antibodies showed binding activity to the native forms of GP expressed on the cell surface or the mucin-like region of GP, but not the denatured form of GP. They also reacted specifically with GP, and remained reactive to GP at the lowest tested concentrations ranging from 1.953 to 7.8 ng/mL. However, none of the antibodies neutralized Ebola virus infection in vitro, suggesting that these IgG antibodies are unable to block Ebola virus infection. Thus, these IgG antibodies possess GP-binding activity but not Ebola virus-neutralizing activity.
Go to : 
Materials and Methods
Animals and cells
Six-week-old female BALB/c mice were purchased from Daehan Biolink (Eumseong, Korea). The mice were cared for under the guidelines of the Kangwon Institutional Animal Care and Use Committee-approved protocols (KW-130419-1). Sp2/0-Ag14 cells were purchased from Korean Cell Line Bank (Seoul, Korea). HEK293 cells, a human embryonic kidney cell line, were kindly provided from JH Lee (Kangwon National University, Korea). The cells were maintained in cDMEM media (Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum [FBS], 1% L-glutamine and 1% penicillin/streptomycin).
DNA vaccines coding for Ebola virus GPs and recombinant GPs
A GP DNA vaccine, pcDNA3-GP was previously tested [10]. It encodes GP of Zaire Ebola virus strain Gabon-94 (GenBank No. U77384.1). Mammalian cell-derived recombinant GP was purchased from Y-Biologics (Daejeon, Korea). Escherichia coli-derived GP1 proteins (GP1a, GP1b, GP1c, GP1d, and GP1e) were previously produced and tested [10].
Stable DNA transfection
HEK293 cells were transfected with 2 µg of pcDNA3-GP using JetPEI transfection reagents (Polyplus-transfection Inc., New York, NY, USA). One day after DNA transfection, the cells were treated with G418 at the concentration of 400 µg/mL.
Delivery of GP DNA vaccines and GP
For intramuscular (IM)-electroporation (EP) delivery of DNA vaccines, mice were injected intramuscularly with 50 µg of pcDNA3-GP per mouse in a final volume of 50 µL of phosphate-buffered saline (PBS) using a 31-gauge needle. The injections were followed by EP at 0.2 volts for 4 seconds using Cellectra of GeneOne Life Science (Seoul, Korea) in accordance with the manufacturer's protocol. Plasmid DNA was produced in bacteria and purified using endotoxin-free Qiagen kits according to the manufacturer's protocol (Qiagen, Valencia, CA, USA). For delivery of recombinant GP, mice were injected intraperitoneally with 20 µg of GP per mouse emulsified in 400 µL of incomplete Freund's adjuvant. Mice were also injected intravenously with 20 µg of recombinant GP per mouse in a final volume of 200 µL of PBS.
Production of hybridomas displaying GP-binding activity
Mice were sacrificed 3 days after the last injection and their splenocytes were prepared for fusion. The fusion was performed as previously described [9]. Briefly, the splenocytes (7×107 cells) were fused with 2×107 Sp2/0-Ag14 cells using 50% polyethylene glycol 4000 (PEG4000). The cells were incubated at 37℃ for 10 days in cDMEM containing hypoxanthine-aminopterin-thymidine (HAT). Subsequently, the cells were incubated with cDMEM containing hypoxanthine-thymidine (HT). After 15 days, the cell supernatants were screened for Ab-secreting hybridomas using enzyme-linked immunosorbent assay (ELISA). For production of MAbs, the Ab-secreting hybridomas were diluted to make less than a single cell per well using the limiting dilution sub-culture method [11]. PEG4000, HAT and HT were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Purification of MAbs
Hybridoma cells were cultured in cDMEM containing HT. The cell supernatants were collected and used for purification of MAbs. MAbs (IgG) were purified using the protein G resin column in accordance with the manufacturer's protocol (Peptron, Daejon, Korea). Briefly, the protein G resin column was pre-equilibrated with 10 mL binding buffer (0.1 M sodium phosphate buffer, 0.15 M NaCl, pH 7.4). Samples were filtered through a 0.2 µm filter and mixed with an equal volume of binding buffer. The mixed samples were loaded onto the resin column, followed by 2 washes with 10 mL of binding buffer. Finally, the bound antibodies were eluted by adding elution buffer (0.2 M glycine buffer, pH 2.5). In particular, 0.5 mL of each eluted sample fraction was added with 65 µL of neutralization buffer (1 M Tris/HCl buffer, pH 9.0) to adjust pH. Finally, the antibodies were dialyzed in PBS.
ELISA
Each well of a 96 well plate was coated at 4℃ overnight with recombinant GP and GP1 proteins (0.5 µg/mL in PBS). The next day the plate was washed 3 times with washing buffer (PBS+0.05% Tween 20) and then blocked for 1 hour with blocking buffer (washing buffer+2% bovine serum albumin). Then, the plate was reacted with test samples for 1 hour. After this, the plates were washed 3 times with washing buffer and reacted for 1 hour with horseradish peroxidase (HRP)-conjugated anti-mouse IgG (H [heavy]+L [light]) diluted in blocking buffer. After washing the plate, an ABTS solution (Merck Millipore, Billerica, MA, USA) was added to each well. Optical density (OD) was read at 405 nm using an ELISA reader. In particular, for the determination of the relative levels of GP-specific IgG subclasses, HRP-conjugated anti-murine IgG1, IgG2a, IgG2b and IgG3 antibodies (Thermo Scientific, Waltham, MA, USA) were substituted for HRP-conjugated anti-murine IgG (H+L). HRP-conjugated anti-mouse IgG (H+L) was purchased from Komabiotech (Seoul, Korea).
Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and Western blot assay
Hybridoma cell supernatants or antibody samples were separated on a 12% SDS-polyacrylamide gel. The proteins on the gel were analyzed by staining with the brilliant blue R staining solution (Elpis-Biotech, Daejon, Korea). The proteins were also electrophoretically transferred to Immobilin-P membranes (Merck Millipore). The membrane was pre-equilibrated with TBST solution (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, and 0.1% Tween 20) containing 5% skim milk for 1 hour. For detection of antibodies, the membrane was incubated with HRP-conjugated anti-mouse IgG (H+L) (Komabiotech) for 1 hour at room temperature. In each step, the membrane was washed 3 times with TBST. The immunoreactive protein bands were visualized using the ECL detection reagents (PhileKorea, Seoul, Korea). In one case, HEK293-GP cell lystes were separated on a 10% SDS-polyacrylamide gel, and transblotted to Immobilin-P membranes. The membrane was cut into many strips, which were reacted with each of the purified IgG and, as a control, anti-Ebola virus GP commercial antibodies raised in rabbits (Sino Biologicals, Beijing, China). This was followed by incubation with HRP-conjugated anti-mouse IgG (H+L) or HRP-conjugated anti-rabbit IgG (Komabiotech) for Western blot assay.
Flow cytometry
HEK293-GP or HEK293 cells (5×105) were reacted at 4℃ for 30 minutes with 2 µL of sera or 1 µg of the purified IgG antibodies in fluorescence-activated cell sorting (FACS) buffer (PBS+1% bovine serum albumin). This was followed by reaction at 4℃ for 30 minutes with fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG (whole molecule) or anti-mouse IgG (Fc). In each step, the cells were washed twice with FACS buffer. The FITC-conjugated anti-mouse IgG (whole molecule) and anti-mouse IgG (Fc) were purchased from Sigma-Aldrich and Abcam (Cambridge, UK), respectively. Finally, the cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences, San Diego, CA, USA).
Ebola virus neutralizing assay
Vero E6 cells were cultured in each well of a 96-well tissue culture plate. The following day, when the cells were approximately 80% confluent, 100 µg/mL of each of the 5 antibodies, as well as 2G4 used as a positive control for neutralization [6], were serially diluted in cDMEM. To this, an equal volume of Ebola virus Makona (GenBank No. KJ660348.2) was added so that each well would receive 100 plaque forming units. The virus-antibody mixture was incubated for 1 hour and then 100 µL of the virus-antibody mixture was added to the 96-well. After incubating the 96-well plate for 1 hour at 37℃, the mixture was removed and 200 µL of 2% FBS DMEM was added to each well. The plate was returned to the 37℃ incubator and plates were read for cytopathic effect 13-day post-infection. This assay was performed in the biosafety level 4 laboratory at the National Microbiology Laboratory, part of the public Health Agency of Canada in Winnipeg, Manitoba, Canada.
Go to :
Results
GP DNA immunization and serum reactivity to a native GP antigen
We previously reported that GP DNA vaccines induce Ag-specific antibody responses [10]. In this study, we investigated whether GP DNA vaccines might induce IgG responses to the native forms of GP expressed on the cell surface. Therefore, HEK293 cells were transfected with pcDNA3-GP and then selected against G418. Cells from the six G418-selected cell colonies were reacted with GP-specific immune sera [10], followed by flow cytometry using FITC-conjugated anti-mouse IgG (whole molecule) as the secondary Ab (Fig. 1A). The HEK293-5 cells, which displayed the prominent binding reactivity to the immune sera, were chosen and tested again for their GP expression status using Western blot assay (Fig. 1B). As seen in Fig. 1B, HEK293-5 displayed 160-140 kDa GP bands, as opposed to control HEK293 cells. This cell was designated HEK293-GP. We next immunized mice by IM-EP with pcDNA3-GP at 0 and 4 weeks and then bled at 6 weeks. The sera were collected and reacted with HEK293-GP cells. As shown in Fig. 1C, the immune sera reacted with HEK293-GP cells, but not with HEK293 cells (without GP expression) by flow cytometry using FITC-conjugated anti-mouse IgG (Fc) as the secondary Ab. This result shows that GP DNA vaccines can indeed induce IgG antibodies that recognize the native forms of GP expressed on the cell surface in vivo.
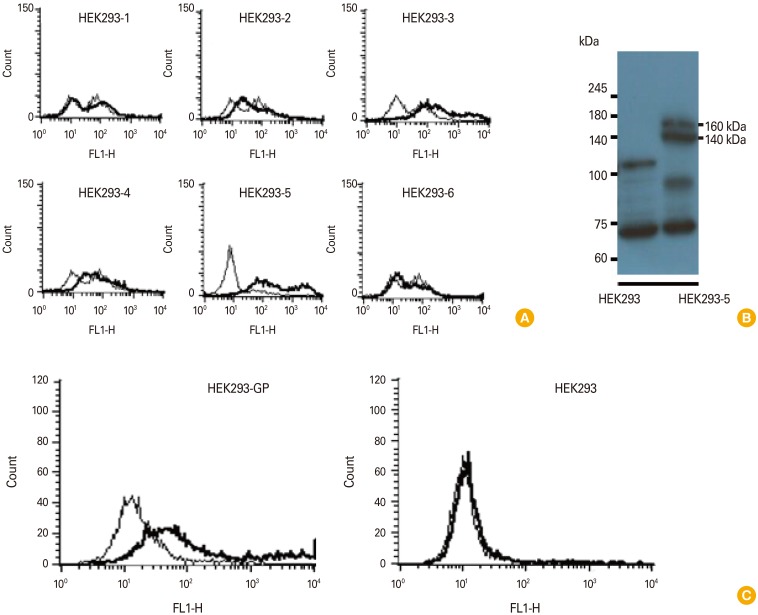 | Fig. 1Stable transfection of HEK293 cells with pcDNA3-GP and selection of GP-expressing HEK293 cells (A, B), and evaluation of sera obtained after GP DNA vaccination for their ability to recognize GP expressed on the cell surface. (A) HEK293 cells were stably transfected with pcDNA3-GP. The G418-selected cell colonies (HEK293-1, 2, 3, 4, 5, 6) were reacted with 2 µL of GP-specific immune sera, followed by reaction with FITC-conjugated anti-mouse IgG (whole molecules) for flow cytometry. The cells were measured for reactivity to GP expressed on HEK293 cells. (B) HEK293-5 cells which prominently reacted with anti-GP sera were further cultured and then the cell lysates were run on a 10% SDS-PAGE gel, transferred to a nitrocellulose membrane and reacted with commercial anti-GP antibodies for Western blot assay. (C) Mice were injected by IM-EP with pcDNA3-GP (50 µg/mouse) at 0 and 4 weeks. The mice were bled at 2 weeks following the final injection and sera were collected. (C) The sera were reacted with 5×105 HEK293-GP and control HEK293 cells, followed by incubating with FITC-conjugated anti-mouse IgG (Fc) for flow cytometry. GP, glycoprotein; FITC, fluorescein isothiocyanate; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; IM, intramuscular; EP, electroporation.
|
Production of monoclonal IgG antibodies and evaluation of their antigen specificity
We previously reported that DNA priming and protein boosting can preferentially generate hybridomas secreting IgG antibodies [10]. To generate IgG antibodies with higher antigen-binding activity, the immunization scheme was modified: mice were injected by IM-EP with pcDNA3-GP (50 µg/mouse) at 0, 4, and 12 weeks, followed by intraperitoneal injection at 20 weeks with recombinant GP (20 µg/mouse) emulsified in incomplete Freund's adjuvant. Four weeks later, the mice were intravenously injected with recombinant GP (20 µg/mouse) in PBS. The mice were sacrificed 3 days after the final injection and the splenocytes were used for fusion. In this fusion experiment, 12 hybridomas, the cell supernatants of which displayed 10 times higher OD values than those of the negative control by ELISA and displayed both heavy and light chains of IgG by Western blot assay, were selected (data not included). These 12 hybridomas were designated C36, D11, D12, D34, E2, E8, E12, E22, E31, E49, E60, and E140. For MAb production, the 12 selected hybridomas were diluted to make a single cell clone using the limiting dilution sub-culture method [11]. As a result, 7 monoclonal clones were obtained and designated C36-1, D11-3, D12-1, D34-2, E12-10, E49-3, and E140-2 (Table 1). Unfortunately, we were unable to generate any clones secreting GP-specific IgG antibodies for clones E2, E8, E22, E31 and E60. In addition, we obtained only IgM-secreting single clones (e.g., E12-10 and E49-3) from hybridomas (E12 and E49). Finally, IgG antibodies were purified from the cell supernatants of 5 clones (C36-1, D11-3, D12-1, D34-2, and E140-2) using the protein G column. They all resolved into 2 protein bands (50-kDa H chains and 25-kDa L chains) on a 12% SDS-polyacrylamide gel (Fig. 2A). They also reacted with Ebola virus GP, but not with MERS coronavirus-spike protein and SFTS virus GP, as determined by ELISA (Fig. 2B). This result suggests that these purified IgG antibodies are indeed antigen-specific. Furthermore, IgG antibodies from clones C36-1, D11-3, D12-1, D34-2, and E140-2 remained reactive to GP at the lowest tested concentrations of 1.953, 7.8, 7.8, 1.953, and 1.953 ng/mL, respectively (Fig. 2C). In this case, the lowest tested concentrations are defined as the lowest IgG concentrations showing positive optical density values compared to those of control IgG (2 µg/mL) when IgG samples were serially diluted by 2-fold from the dose (2 µg/mL) and then reacted with recombinant GP in ELISA. When the IgG antibodies from the five clones were further tested for their reactivity to five different GP1 proteins (GP1a, GP1b, GP1c, GP1d, and GP1e), only the IgG antibodies from clone D12-1 reacted with GP1e while the IgG antibodies from clones C36-1, D11-3, D34-2, and E140-2 failed to react with any of the GP1 proteins (Fig. 2D). This result suggests that the IgG antibodies from clones C36-1, D11-3, D34-2, and E140-2 may recognize either the tertiary structure of GP or the GP regions excluding the receptor-binding domain and mucin-like region, and that the IgG antibodies from clone D12-1 might recognize the mucin-like region of GP. This is illustrated in Table 1.
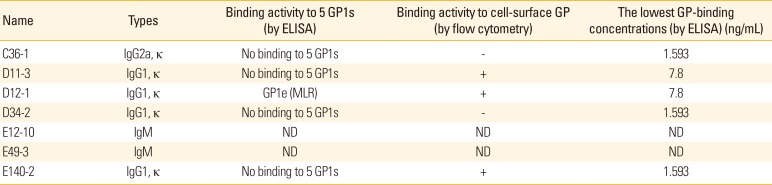
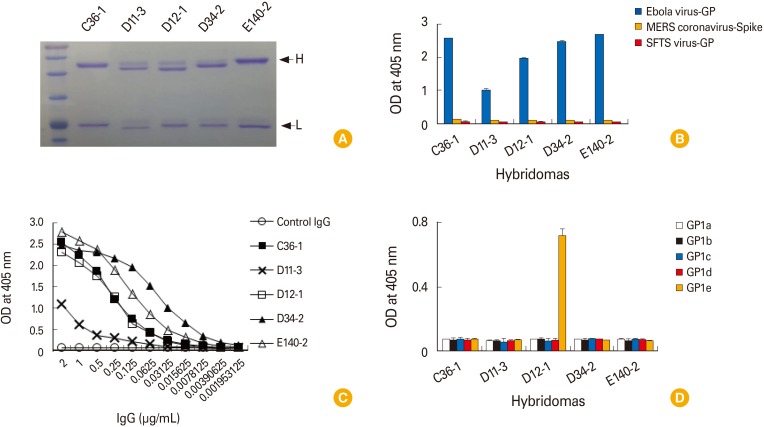 | Fig. 2Purification of the IgG antibodies from the five hybridoma clones, and the evaluation of their Ag-specific binding activity. (A) The hybridomas (D36, D11, D12, D34, and E140) were diluted to make a single cell by the limiting dilution sub-clone method. The resulting monoclonal hybridoma clones were designated C36-1, D11-3, D12-1, and E140-2. IgG antibodies were purified from the cell supernatants using the protein G column. Two µg of the purified IgG antibodies were run on a 12% SDS-PAGE for brilliant blue R staining. (B) The purified IgG (2 µg/mL) were reacted with Ebola virus GP, MERS coronavirus spike protein and SFTS virus GP for ELISA. (C) Evaluation of the lowest GP-binding concentration of IgG antibodies. Two micrograms per milliliter of IgG antibodies from the clones, C36-1, D11-3, D12-1, and E140-2 were serially diluted by 2-fold and then reacted with GP in parallel with control IgG (2 µg/mL) to determine the lowest GP-binding concentration using ELISA. (D) Five recombinant GP1 proteins (1 µg/mL) were coated and reacted with the five purified IgG antibodies (0.2 µg/mL) from clones, C36-1, D11-3, D12-1, and E140-2 for ELISA. SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel; GP, glycoprotein; MERS, Middle East respiratory syndrome; SFTS, severe fever with thrombocytopenia syndrome; ELISA, enzyme-linked immunosorbent assay; OD, optical density.
|
Table 1
The binding properties of the purified IgG and IgM antibodies from 7 hybridoma clones
![]()
The ability of the monoclonal IgG antibodies from clones (C36-1, D11-3, D12-1, D34-2, and E140-2) to react with the native forms of GP expressed on the cell surface
We next tested whether the IgG antibodies from 5 clones (C36-1, D11-3, D12-1, D34-2, and E140-2) might react with the native forms of GP expressed on the surface of HEK293-GP cells. For this testing, 1 µg of the purified IgG was reacted with HEK293-GP cells, as well as control HEK293 cells without GP expression, followed by flow cytometry. As shown in Fig. 3, the IgG antibodies from clones D11-3, D12-1, and E140-2 showed positive reactivity to HEK293-GP cells but not to HEK293 cells. In contrast, the IgG antibodies from clones C36-1 and D34-2 displayed no reactivity to HEK293-GP cells. This result suggests that the IgG antibodies from clones D11-3, D12-1, and E140-2 may recognize the native forms of GP expressed on the cell surface.
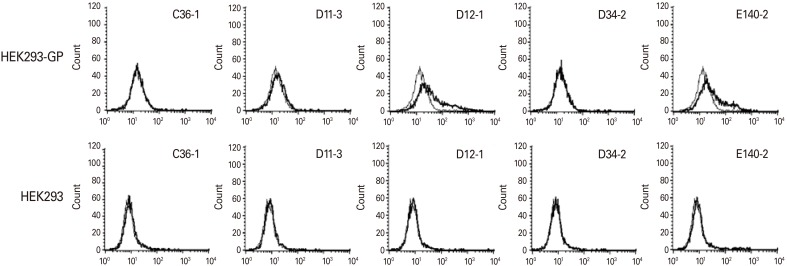 | Fig. 3Evaluation of the five purified IgG antibodies for their binding activity to the native forms of GP expressed on the cell surface. One microgram per mililiter of the IgG antibodies from clones, D36-1, D11-3, D12-1, D34-2, and E140-2 were reacted with 5×105 HEK293-GP and HEK293 cells, followed by incubation with FITC-conjugated anti-mouse IgG (Fc) for flow cytometry. Thin line: control IgG, Thick line: experimental IgG. GP, glycoprotein; FITC, fluorescein isothiocyanate.
|
A lack of binding activity of the 5 purified monoclonal IgG antibodies to the denatured form of GP
We next tested whether the IgG antibodies from 5 clones (C36-1, D11-3, D12-1, D34-2, and E140-2) might react with the denatured form of GP. As shown in Fig. 4, none of the IgG antibodies recognized the denatured forms of GP by Western blot assay. However, commercial antibodies as a control recognized the denatured forms of GP. This result suggests that the five purified IgG antibodies are unable to recognize the denatured forms of GP.
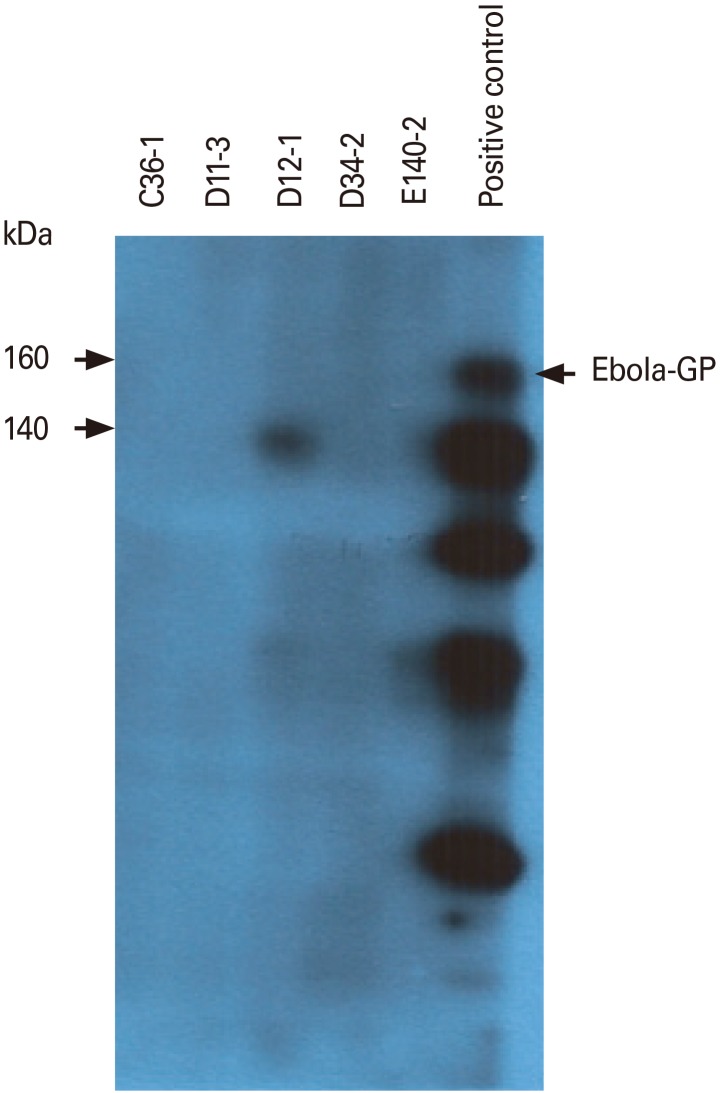 | Fig. 4Binding activity of the five purified IgG antibodies to the denatured forms of GP by Western blot assay. Thirty micrograms of HEK293-GP cell lysates were separated on a 10% sodium dodecyl sulfate-polyacrylamide gel. The proteins on the gel were transferred to a nitrocellulose membrane. The membrane was cut into strips, which were reacted with each of the five purified IgG antibodies (1 µg/mL) or commercial anti-Ebola virus GP, followed by Western blot assay. Arrows indicates Ebola virus GP. GP, glycoprotein.
|
A lack of Ebola virus neutralization activity of the 5 IgG antibodies
We also investigated whether the IgG antibodies from 5 clones (C36-1, D11-3, D12-1, D34-2, and E140-2) might possess the ability to neutralize Ebola virus infection. As shown in Table 2, none of the IgG antibodies neutralized Ebola virus infection. However, a control antibody (2G4) neutralized Ebola virus even at the concentration of 3.125 µg/mL. This result suggests that the 5 monoclonal IgG antibodies are unable to neutralize Ebola virus infection and that these antibodies may be of no use for controlling Ebola virus infection.
Table 2
Ebola virus neutralization activity of the 5 purified monoclonal IgG antibodies
![]()
Go to :
Discussion
In this study, we observed that vaccination with DNA vaccines encoding cell membrane-targeting viral antigens led to induction of IgG antibodies recognizing the native forms of the antigens expressed on the cell surface. For instance, immune sera, displaying reactivity to GP expressed on the cell surface, were demonstrated by flow cytometry using both FITC-conjugated IgG (Fc) and FITC-conjugated IgG (whole molecule). In this case, FITC-conjugated IgG (Fc) recognized only IgG types, as opposed to FITC-conjugated IgG (whole molecule) (data not included). Moreover, DNA vaccines coding for cell membrane-associated antigens were reported to be more effective at inducing Ag-specific immune responses, as compared to those coding for intracellular antigens [12]. These data including ours suggest that DNA vaccines encoding any viral surface membrane proteins can induce IgG responses to the native and outer-membrane forms of the viral proteins. Thus, it is possible that these IgG antibodies may be utilized to target viral surface antigens as either diagnostic or neutralizing antibodies. Previously, we reported that DNA vaccination alone resulted in the preferential production of IgM-secreting hybridomas over IgG-secreting hybridomas [10]. In that study, use of recombinant proteins in the DNA vaccination platform reversed this propensity, highlighting the notion that proteins may be a useful vaccine type for increased production of IgG-specific hybridomas in DNA vaccination. In the current study, we successfully generated IgG-secreting hybridomas by boosting the GP DNA vaccine-primed mice with GP emulsified in incomplete Freund's adjuvant and finally with GP dissolved in PBS. This result corroborates the notion that proteins, as a booster vaccine type, may play a critical role in boosting the production of IgG-secreting hybridomas in the DNA vaccine platform.
In the screening scheme using ELISA and Western blot assay, we were able to obtain a total of 12 polyclonal hybridomas secreting GP-specific IgG types. Among these, we obtained five clones (producing an IgG isotype) and two clones (producing an IgM isotype) by the limiting dilution sub-culture method. The purified monoclonal IgG antibodies from five clones (C36-1, D11-3, D12-1, D34-2, and E140-2) reacted with Ebola virus GP, but not MERS coronavirus Spike protein and SFTS virus-GP, suggesting that they are indeed antigen-specific. Moreover, they were IgG kappa types and remained reactive to Ebola virus GP at the lowest concentrations ranging from 1.593 to 7.8 ng/mL. In particular, the IgG antibodies from 3 clones (D11-3, D12-1 and E140-2) recognized GP expressed on the cell surface of HEK293-GP, suggesting that they may recognize the native and outer-membrane forms of GP. On the other hand, the IgG antibodies from one clone (D12-1) recognized GP1e (containing mucin-like region) alone, suggesting that this IgG antibody recognizes the mucin-like region of GP. However, the IgG antibodies from four clones (C36-1, D11-3, D34-2, and E140-2) recognized none of the five recombinant GP1s, suggesting that the four purified IgG antibodies may recognize the tertiary forms of GP and/or other GP sites excluding the mucin-like region and receptor-binding domain of GP. Moreover, none of the five purified IgG antibodies recognized the denatured forms of GP. These data are in line with our observation that sera from animals immunized with GP DNA vaccines were unable to recognize the denatured forms of GP by Western blot assay (data not included). Taken together, these data show that the five purified IgG antibodies can recognize only a native from of GP. Here, it is notable that the remaining five polyclonal hybridomas (E2, E8, E22, E31, and E60) appear to lose their ability to produce IgG antibodies after continuous cell culture and passage, leading to a failure in making an IgG producer. Among numerous single clones obtained after limiting dilution sub-culture from the polyclonal hybridomas, not a single clone was observed to secrete Ag-specific IgG antibodies (data not included). In this context, it is highly likely that the loss of IgG productivity might be ascribed to a decrease in the fraction of IgG-producing hybridoma cells over time, possibly resulting from the loss of the chromosomes containing the gene loci for the IgG heavy and light chains. This is supported by the previous finding of others [13]. On the other hand, clones E12-10 and E49-3, displaying the highest OD values among numerous clones, were secreted as an IgM producer. Prior to limiting dilution sub-culture, two hybridomas (E12 and E49) generated IgG types, but later lost their ability to generate IgG antibodies. It is likely that in the case of 2 hybridomas (E12 and E49), IgG-producing cell populations may have disappeared and then been replaced by IgM-producing cell populations. This notion is supported by our observation that the cell supernatants from two hybridomas (E12 and E49) exhibited high binding activity to all five different recombinant GP1s (suggestive of antigen-non-specific binding) in an ELISA (data not shown). In addition, these cell supernatants also reacted with GP in an ELISA using HRP-conjugated IgM as well (data not shown), confirming that clones E12-10 and E49-3 are indeed from polyclonal cells producing IgG and IgM antibodies. It has been reported that Ebola virus GP is responsible for binding to the host cell's receptor for viral entry [45]. Ebola virus GP-specific antibodies including 2G4 monoclonal IgG antibodies are known to block viral infection to the host cells [6]. In this study, we observed that the five purified IgG antibodies were unable to block Ebola virus infection to the host cell in vitro. These data suggest that the five purified GP-specific IgG antibodies including three (recognizing a native GP expressed on the cell surface) do not have any neutralizing activity toward Ebola virus infection.
In summary, we found that among 12 GP-specific IgG-producing hybridomas, five were successfully sub-cloned to IgG-producing single clones. All of the five purified monoclonal IgG antibodies recognized GP in an antigen-specific and IgG dose-dependent manner. In particular, three recognized the native forms of GP expressed on the cell surface and remained reactive to GP at the lowest tested concentrations ranging from 1.953 to 7.8 ng/mL. However, these antibodies were unable to neutralize Ebola virus, suggesting that none of these purified antibodies can block Ebola virus infection. Taken together, the five purified IgG antibodies possess Ebola virus GP-binding activity but not Ebola virus neutralizing activity.
Go to :
 XML Download
XML Download