

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
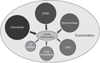
Chronic obstructive pulmonary disease (COPD) is defined as a common, preventable, and treatable disease that is characterized by persistent respiratory symptoms and airflow limitation caused by exposure to noxious particles or gases. The progressive airflow limitation is attributed to chronic inflammation leading to small airway obstruction and parenchymal tissue destruction1. Exacerbation is defined as an acute worsening of respiratory symptoms1 that may be associated with the increase of airway inflammation mediated by neutrophils; lymphocytes; eosinophils; and the associated mediators, such as interleukin-8 (IL-8), regulated on activation, normal T cell expressed and secreted, and neutrophils elastase2. The causes of exacerbation are complex and mainly include respiratory, viral, or bacterial infections, and environmental pollution3, which lead to an increase in inflammatory burden and a decrease in protective defense by host immunity. In the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) study4, which is a clinical observation on COPD patients over a period of 3 years, the exacerbation frequency appeared to be linked to prior history of exacerbation and severity of diseases. COPD patients with frequent exacerbations during the previous year tended to have higher susceptibility to exacerbations in the following years. What is the underlying mechanism to explain this frequent-exacerbation phenotype of COPD? What is breach of stability of airway inflammation and what triggers exacerbation? The co-existing eosinophilic inflammation and concomitant chronic diseases will be discussed (Figure 1).
Inflammation and Exacerbation
1. Airway inflammation
The airflow limitation in COPD is associated with abnormal inflammatory response. Progression of airflow limitation is associated with the increase in accumulation of mucus exudates in the lumen and infiltration of the small airway wall by inflammatory cells5. A variety of inflammatory cells are present in the airways of patients with COPD. Neutrophils, macrophages, eosinophils, mast cells, and CD8+ lymphocytes have been shown to play important roles in the inflammatory processes in COPD6789.
Neutrophils are one of the major inflammatory cell types in COPD5. Increased numbers of neutrophils in sputum were found to be correlated with a rapid decline in patients' forced expiratory volume in one second (FEV1) in a 15-year period of observation10. In stable COPD patients, FEV1 showed a significant inverse correlation with the number of neutrophils in induced sputum11 and in the sub-epithelium from bronchial biopsies12, suggesting that neutrophilic inflammation of the airways may play an important role in the pathogenesis of COPD.
Macrophages can be activated by cigarette smoke to release inflammatory mediators and matrix metalloproteases (MMPs). The number of alveolar macrophages was correlated to the extent of emphysema formation13 and to the severity of airway obstruction12. The MMPs, Zn2+-dependent proteases, and Ca2+-dependent proteases can degrade a broad range of connective tissue proteins and are involved in remodeling damaged tissue. Increased activity of MMP-8 (collagenase) and MMP-9 (gelatinase) was detected in induced sputum from patients with COPD, and this activity was correlated to the degree of airflow limitation14. In the animal model of knockout mice, MMP-12 deficiency can block the increase in macrophages in the lungs and the development of emphysema in response to long-term exposure to cigarette smoke15. In addition, MMP-12 mediates smoke-induced inflammation by releasing tumor necrosis factor α (TNF-α) from macrophages with subsequent endothelial activation, neutrophil influx, and proteolytic matrix breakdown caused by neutrophil-derived proteases16.
The role of eosinophils in COPD and the mechanism of eosinophil influx into airways remain to be clarified. Hogg et al.5 reported that eosinophils do exist in the small airways of various severity of COPD. In a prospective clinical observation, nearly one third of COPD patients had sputum eosinophilia and the number of eosinophils was significantly correlated to the level of exhaled nitric oxide17. The degree of eosinophilic inflammation has been related to early changes in lung function and smoking habits. The higher counts of eosinophils in induced sputum is associated with higher pack-years and lower peak expiratory flow 25%–75% values18. In a retrospective analysis, FEV1 reversibility was weakly correlated with the sputum eosinophil level. Patients with FEV1 >0.4 L and >15% increment had higher sputum eosinophil levels whereas the level did not differ when dichotomized by FEV1 increment >0.2 L and >12%19. The response to corticosteroids in COPD patients were better in a subset of patients presenting with more eosinophils and higher levels of eosinophil cationic protein20, suggesting that corticosteroids may be effective especially for patients with COPD with eosinophilic airway inflammation2122. In addition, we had reported that inhaled fluticasone (200 µg/day) can significantly suppress the numbers of eosinophils in the airways of COPD. Suppression of such eosinophilic inflammation was not associated with improvement in lung function. Brightling et al.23 also reported that the improvement in FEV1 is not associated with a reduction in the sputum eosinophil count in COPD with sputum eosinophilia.
Increased CD8+ T lymphocytes have been shown in the central and peripheral airways and lung parenchyma in COPD12132425. The increased number of CD8+ T lymphocytes in the subepithelium has been correlated with the severity of airflow limitation26. Increased cytotoxic activity of CD8+ T cells was observed in induced sputum of patients with COPD compared to those of smokers without COPD and healthy subjects. The expression of perforins, a secreted molecule that causes cytolysis, was also high27. However, one in vitro study demonstrated that cytolytic activity of CD8+ T cells against alveolar epithelial cells is perforin-independent and is attributed entirely by TNF-α, which is expressed on CD8+ T cells28. However, the contribution of CD8+ T cell and the ratio of CD4+/CD8+ cells to the pathogenesis of COPD as well as the cell recruitment to the airways are not clear.
2. Acute exacerbation
Acute exacerbation is associated with increased inflammatory burden leading to worsening respiratory symptoms including dyspnea and sputum production. In the ECLIPSE study, the exacerbation rates were 0.85, 1.34, and 2.0 (number/yr/patient) for patients with Global Initiative for Obstructive Lung Disease (GOLD) stages 2, 3, and 4, respectively, during the first year of follow-up4. The exacerbation rates were significantly associated with post-bronchodilator FEV1, impairment in health status, history of reflux or heartburn, white blood cell count, and prior history of exacerbation. In addition, acute exacerbation of COPD had been found to be an independent factor of mortality, suggesting more exacerbations with higher mortality29. In a subgroup analysis in the Inhaled Steroids in Obstructive Lung Disease in Europe (ISOLDE) study, 39% and 36% of patients with or without inhaled corticosteroid treatment had no exacerbation in the first year of follow-up. During the three-year study period, 22% and 23% of patients respectively had no exacerbations30. In a retrospective observational study, 85.5%, 80.4%, 60.6%, and 48.1% of COPD patients with GOLD classification group A, B, C, and D had no acute exacerbation in the previous year before enrollment31.
What Is the Breach of Stability of Airway Inflammation?
1. Bacterial and viral infection
In a prospective observational study, 55% and 29% of COPD patients had bacteria or virus-associated exacerbations, respectively32. The most common causes of bacteria-associated exacerbations were Haemophilus influenzae, Moraxella catarrhalis, and Streptococcus pneumoniae33. The major viruses associated with COPD exacerbations were rhinovirus (common cold), coronavirus, influenza, and parainfluenza34. Papi et al.35 reported that patients with infectious exacerbation (bacteria, virus, or coinfection) may have longer hospitalization and greater impairment of lung function compared to those non-infectious exacerbations. Sputum neutrophilia were observed in all exacerbations and was associated with disease severity. Sputum eosinophilia was observed during virus-associated exacerbations35.
Normally, the lower airways are sterile. Bacterial components such as flagella, lipoproteins, and lipopolysaccharides may initiate inflammatory reactions by releasing a variety of pro-inflammatory mediators and recruiting inflammatory cells36. For example, protein A from staphylococci can recognize TNF-α receptors in airway epithelium and induce production of IL-8, which is an important chemokine for neutrophils37. Pseudomonas aeruginosa flagella can activate airway epithelial cells to produce IL-8 through toll-like receptors 2 and 538. In stable COPD patients, sputum neutrophils and IL-8 levels were higher than those in healthy subjects, which suggested ongoing neutrophilic inflammation in the airways11. Sputum IL-8 levels were reported to be correlated with total bacterial count, and bacterial load in the airways was associated with increased exacerbation frequency39. Rhinovirus infection can increase the production of IL-8, IL-6, and granulocyte-macrophage colony-stimulating factor (GM-CSF) from airway epithelial cells; IL-8 production was associated with viral replication40. In a study in mice, influenza virus infection resulted in influx of neutrophils, monocytes, lymphocytes, and eosinophils as well as various chemokines and cytokines 3 to 10 days after infection41.
2. Air pollution and temperature
Increased concentration of outdoor pollutants such as NO2, O3, SO2, particulate matter 2.5 (PM2.5), and particulate matter 10 (PM10) were associated with increased respiratory symptoms, acute exacerbations, and hospital admissions in COPD patients42. Inhalation of high concentration of NO2 may correlate to acute epithelial damage43. Cultured human bronchial epithelial cells exposed to NO2 could increase the expression of IL-1β, IL-8, GM-CSF, and TNF-α44. SO2 can lead to airway edema, inflammation, and fibrosis in an animal model after 3–25 days' exposure to different levels of SO245. O3 and PM10 have been reported to increase the incidence of hospital admission for pneumonia and acute exacerbation of COPD in a meta-analysis based on city-specific regression models46. Short-term PM2.5 exposure was associated with an increased risk of about 0.9% for COPD hospitalization47. In an epidemiologic study, for every 10 µg/m3 increase in O3, NO2, and SO2 concentrations, the risks of hospitalization for COPD increase to about 0.58%, 0.38%, and 0.44%, respectively48. Seasonal variations and temperature may affect the incidence of acute exacerbation of COPD. Tseng et al.49 reported that a 1℃ decrease in air temperature was associated with a 0.8% increase in the exacerbation rate on event-days. The cold temperature was reported to be linked to bronchoconstriction50 and FEV1 decrease51 especially for COPD patients.
3. Gastroesophageal refluxate
The gastroesophageal refluxate contains gastric acids, bile, and pepsin. Gastroesophageal reflux (GER) is a major cause of subacute and chronic cough. In a questionnaire-based prospective survey, the prevalence of GER disease (GERD) was 26.7% among stable COPD patients, and severe GERD symptoms were associated with more frequent exacerbations of COPD and hospitalization with crude relative risks of 3.42 and 3.66, respectively51. Casanova et al.52 reported that severe COPD patients had high prevalence of GERD (62%) confirmed by esophageal 24-hour pH monitoring but 58% of them did not have any reflux symptoms. In addition, the annual rate of exacerbations of COPD was twice as high in patients with GER symptoms compared to those without GER symptoms (3.2/yr vs. 1.6/yr, p=0.02)53. In our previous study, the amount of total bile acids in induced sputum was significantly higher in patients with GER and asthma-associated GER symptoms compared to that of healthy control subjects (p<0.005). Bile acid significantly induced transforming growth factor β1 (TGF-β1) production from airway epithelial cells, which in turn enhanced fibroblast proliferation54 and altered alveolar epithelial permeability, which may contribute to the pathogenesis of lung injury55. Airway epithelium exposed to bile acids could induce fibrosis via production of connective tissue growth factor (CCN2). CCN2 is essential for TGF-β1–induced fibrogenesis and functions as a downstream mediator of TGF-β1 action on fibroblasts56. The major bile acid, chenodeoxycholic acid, stimulated alveolar epithelial cells to increase IL-8 production through p38 and c-Jun N-terminal kinase activation. This may enhance neutrophilic inflammation in chronic airway disorders57. How to intervene and attenuate GER-related inflammation and fibrogenesis in chronic airway disorders is worth further investigation.
4. Bronchiectasis and bacterial colonization
The prevalence of bronchiectasis in COPD varies according to COPD severity. In a prospective observational study, 50% of moderate to severe COPD patients had significant detectable bronchiectasis on high-resolution computed tomography scanning. Lower lobe bronchiectasis was associated with bacterial colonization quantified by sputum sampling58. Martinez-Garcia et al.59 reported that the prevalence of bronchiectasis in moderate and severe COPD patients was 34.7% and 72.5%, respectively. Patients with bronchiectasis had more chronic expectoration, more exacerbation indices, more severe airflow obstruction, and greater positive cultures of potentially pathogenic microorganism. Haemophilus influenzae and Pseudomonas aeruginosa were the most common microorganisms isolated from sputum59. In addition, moderate-to-severe COPD patients with bronchiectasis was associated with increased all-cause mortality (37.4%) compared to COPD patients without bronchiectasis (9.3%)60.
5. Asthma and eosinophilic inflammation
Eosinophilic airway inflammation is a characteristic feature of atopic asthma and exists in a subgroup of COPD. Bafadhel et al.32 reported that 28% of cases of acute exacerbation of COPD were associated with sputum eosinophilia. In our previous study, 32% of naïve COPD patients had sputum eosinophilia and tended to have higher serum IgE levels and positive allergen tests17. Patients with asthma-COPD overlap syndrome may present with more respiratory symptoms, worse lung function, and increased risk of exacerbation and hospitalization (prevalence ratio, 2.11 and 4.11, respectively) compared to patients with COPD alone61. Hardin et al.62 reported that patients with asthma and COPD overlap syndrome (ACOS) were more likely to experience frequent exacerbations (odds ratio, 3.55) and have had a severe exacerbation in the past years. In a prospective randomized study, a reduction in eosinophilic airway inflammation was associated with a reduction in severe exacerbation of COPD63. Sputum eosinophilia and high blood eosinophil counts are associated with increased responsiveness to steroids in COPD patients22236465. Accordingly, the detection of eosinophilic inflammation is a crucial step to make a precise treatment in COPD patients.
6. Obstructive sleep apnea
In an epidemiologic study for COPD, the risk of obstructive sleep apnea (OSA) assessed by questionnaire was 29.5% among COPD patients66. The prevalence was reported to be 65.9% for moderate to severe COPD confirmed by polysomnography67. Marin et al.68 showed that COPD patients with OSA overlap syndrome without continuous positive airway pressure (CPAP) treatment had higher mortality rates and were more likely to have a severe COPD exacerbation leading to hospitalization (relative risk, 1.70; 95% confidence interval [CI], 1.21–2.38) compared to patients with COPD alone. OSA exacerbated airway inflammation by increasing neutrophil proportion and levels of IL-8 and TNF-α in bronchoalveolar lavage fluid samples from moderate to severe COPD patients. The augmentation of inflammation can be curbed by treatment with CPAP69. The mechanisms of how OSA exacerbates airway inflammation in COPD remain to be elucidated.
The significant predictors or factors that are associated with increased risk of acute exacerbation in COPD is summarized in Table 1.
How to Prevent Exacerbation
1. Airway inflammation
Long-acting bronchodilators such as long-acting muscarinic antagonist (LAMA) and long acting β2-adrenergic receptor agonist (LABA) are the major pharmacological drugs for COPD to improve airflow limitation, respiratory symptoms, and quality of life as well as reduce exacerbations7071. The mechanisms of long-acting bronchodilators in reducing exacerbation either by improving hyperinflation or targeting inflammation remain controversial. The combination of LABA and corticosteroids can reduce the numbers of macrophages72, neutrophils73, and CD8+ T lymphocytes7273 in bronchial biopsy tissue and in induced sputum from patients with COPD. We had reported that the combination of inhaled salmeterol and fluticasone caused a significant reduction in IL-8 and MMP-9 in induced sputum from COPD patients compared to levels in those who received treatment with tiotropium alone74. Tiotropium can inhibit airway inflammation and remodeling75 and attenuate neutrophilic inflammation as well as associated mediators LTB4, IL-6, monocyte chemoattractant protein-1, macrophage inflammatory protein (MIP)-1, MIP-2, and TNF-α in the animal model76. In clinical studies, the odds ratio for tiotropium in reducing exacerbations compared to placebo was 0.74 (95% CI, 0.66–0.83) and hospitalizations 0.69 (95% CI, 0.55–0.87)77. Indacaterol and formoterol can suppress neutrophil-associated mediators such as reactive oxygen species, LTB4, and elastase in a dose-dependent manner78. In a prospective, randomized, blinded, and double-dummy study, it was found that tiotropium was superior to indacaterol in terms of annualized exacerbation rate (0.61 vs. 0.79) for severe COPD79. A combination of indacaterol and glycopyrronium significantly reduced the annual rate of moderate to severe exacerbations versus glycopyrronium alone by 12%, suggesting that the combination of indacaterol/glycopyrronium was superior in preventing moderate to severe exacerbations compared to glycopyrronium alone80. Roflumilast, a]' phosphodiesterase-4 (PDE4) inhibitor, has been reported to significantly reduce moderate to severe exacerbations by 17% compared to placebo for severe COPD patients with chronic bronchitis and frequent exacerbation phenotype81. PDE4 inhibitors were associated with a reduced likelihood of COPD exacerbation with odds ratio 0.77 (95% CI, 0.71–0.83) in a pooled analysis82. Choosing the optimal LABA, LAMA, dual bronchodilator, PDE4 inhibitor, inhaled corticosteroids, or specific combination of different drugs for COPD patients with various severity and phenotype is challenging in our clinical practice.
2. Bacterial and viral infections
Pneumococcal and influenza vaccinations have been recommended for patients with COPD as a preventive strategy1. A recent meta-analysis showed that pneumococcal vaccination can provide significant protection against community-acquired pneumonia (CAP) and reduce the likelihood of a COPD exacerbation83. By contrast, Sehatzadeh84 reported that the time to the first episode of CAP due to pneumococcus or of unknown etiology was not significantly different between the vaccine and control groups. Nevertheless, hospital admission rates and median lengths of hospital stay were lower in the vaccine group but were not statistically significant84. For influenza vaccination, it was found that the vaccine group had fewer episodes of influenza-related acute respiratory illnesses in all severities (mild, moderate, and severe) of COPD, fewer hospitalizations, and fewer requirements for mechanical ventilation84.
1) Air pollution
It is necessary to promote and conduct research on ambient air pollution as a COPD risk factor. It is necessary to form a partnership with government institutions such as the Environmental Protection Agency (EPA) and academic institutions to investigate the detrimental effect of environmental exposure and occupational hazards as well as to apply prevention strategy and patient education. The U.S. EPA has recommended that time and intensity of outdoor activity should be regulated by air quality index since 2009. The N95 respirator was reported to be able to provide excellent protection against airborne particles85.
3. Gastroesophageal refluxate
Aspiration of gastroesophageal refluxate can trigger exacerbation of COPD and enhance airway inflammation and fibrosis as we discussed earlier. Among COPD patients with GERD, those who did not use acid inhibitory treatment regularly had a significantly increased risk of COPD exacerbation with a hazard ratio (HR) of 2.7 as compared to those who were treated regularly with an HR of 1.286. In moderate to severe asthma patients with GERD and nocturnal respiratory symptoms, taking proton pump inhibitors significantly improved morning and evening peak expiratory flow over those taking palcebo87. The ability to identify COPD patients who are at risk for GERD or GER-related chronic cough is essential to COPD management. It is worth conducting randomized trials to investigate the efficacy of anti-acid and anti-reflux therapy on COPD patients with GERD and whether these treatments can decrease respiratory symptoms, improve lung function, and quality of life as well as reduce exacerbation.
4. Bronchiectasis and bacterial colonization
In a prospective study, the incidence of bacterial colonization in bronchiectasis was as high as 64%. The risk factors included diagnosis of bronchiectasis before the age of 14 years (odds ratio [OR], 3.92), FEV1 <80% predicted (OR, 3.91) and presence of varicose or cystic bronchiectasis (OR, 4.8)88. Bacterial infection can cause airway inflammation, which can subsequently result in exacerbation. COPD patients with bronchiectasis have more respiratory symptoms, more exacerbations, and worse prognosis. Azithromycin 250 mg taken daily for 1 year, decreased the frequency of exacerbations (HR, 0.73; p<0.001) and improved the quality of life among COPD patients89. The clinical efficacy on hospitalization, urgent care visit, and intubation was similar between both groups and the adverse reaction (hearing impairment) was more common in the azithromycin group. Uzun et al.90 reported that azithromycin 500 mg taken 3 times a week for 12 months resulted in a significant reduction in the exacerbation rate versus placebo (HR, 0.58; p=0.001) among COPD patients who had 3 or more exacerbations in the previous year. A small study demonstrated that inhaled tobramycin reduced the inflammatory impact of Pseudomonas aeruginosa in a multiresistant Pseudomonas-colonized patient with severe COPD after 2-week treatment. A 42% reduction in exacerbation rate was observed in the 6-month follow-up91. The ability to select patients, choose appropriate antibiotics, decide the duration of treatment are the major concerns for treatment strategy for frequent exacerbations of COPD with bacterial colonization.
5. Asthma and eosinophilic inflammation
Whether eosinophilic inflammation in COPD is attributable to ACOS or is a specific phenotype of COPD is still controversial. However, the response of eosinophilic inflammation to treatment would be more important than the delineation of the differences between these two terms. Early detection of characteristic features of asthma or eosinophilic airway inflammation in COPD patients is necessary before starting treatment. Zanini et al.92 reported that COPD patients with bronchial hyper-responsiveness (BHR) had higher sputum eosinophils than those without BHR. The change in FEV1 was positively correlated with sputum eosinophils while patients had bronchial reversibility1992. The prevalence of elevated serum IgE had been reported to be 47.3% in COPD patients93. Serum IgE levels were associated with COPD severity94 and sputum eosinophilia17. The levels of eosinophils in sputum samples were significantly correlated with exhaled nitric oxide level (eNO)17. The eNO, blood eosinophils, and antigenspecific IgE were significantly higher in ACOS than in COPD alone according to a cross-sectional study in Japan95. Blood eosinophils were proposed to be a surrogate marker for airway eosinophilia in stable COPD96 and a marker of response to inhaled corticosteroids97. COPD patients with blood eosinophils ≥2% treated with inhaled corticosteroid/LABA had significantly fewer exacerbations than those treated with LAMA (relative risk [RR], 0.75; p=0.006, INSPIRE study) or placebo (RR, 0.63; p<0.001)98. It is warranted to conduct a prospective study to determine which marker is better in predicting corticosteroid response in COPD with eosinophilic airway inflammation.
6. Obstructive sleep apnea
CPAP is considered to be the standard treatment for OSA patients. The benefit of CPAP treatment on reducing airway inflammation in COPD is barely discussed. CPAP may reduce oxidative stress by decreasing plasma malondialdehyde in OSA99 and significantly reduce urinary norepinephrine levels and blood pressure in patients with severe OSA100. Briefly, OSA may exacerbate airway inflammation in COPD and CPAP is the optimal choice to reverse the inflammatory process so far.
Conclusion
Though airway inflammation in COPD varies based on severity, there is still a gap between inflammation and exacerbation. Bacterial or viral infection, air pollution, GERD, bronchiectasis with bacterial colonization, and eosinophils may breach the airway stability and lead to overwhelming inflammation and acute exacerbation. The application of antiinflammatory agents and vaccines, prevention strategies for air pollution, early detection of GERD, reduction of bacterial load in bronchiectasis, and measurement of biomarkers that can predict response to corticosteroids are becoming the therapeutic goals to minimize the risks of acute exacerbation. We still need more evidence and clinical studies to achieve these endpoints and improve health-related quality of life among COPD patients.

 XML Download
XML Download