

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The Tembusu virus (TMUV), which was classified into the genus Flavivirus, family Flaviviridae, causes a reduction in egg production, ovarian hemorrhage, acute anorexia, diarrhea, and paralysis in ducks and geese at the beginning of the disease. To date, it has been reported that chickens, pigeons, house sparrows, BALB/c mice, and Kunming mice are susceptible to this newly emerging virus [61114183033]. The morbidity rate in ducks is typically high (nearly 100%), and the mortality rate ranges from 5% to 30% due to secondary bacterial infections. TMUV has become a widespread infectious disease in ducks, leading to serious economic loss in the duck industry in China [36].
Flaviviruses include nearly 70 enveloped positive-sense RNA viruses [10]. Many, such as Dengue virus, Yellow fever virus, West Nile virus, Japanese encephalitis virus and Zika virus, can cause serious diseases in humans and animals [2]. The genome of flaviviruses is approximately 11 kb, containing a single open reading frame and encoding a polyprotein. The polyprotein is processed into three structural proteins (capsid [C], membrane [M], and envelope [E]) and seven nonstructural proteins (NS1, NS2a, NS2b, NS3, NS4a, NS4b, and NS5) by viral and cellular proteases [39].
The E protein is a major structural protein of flaviviruses. Crystallography results have revealed that E protein has three separate domains and forms head-to-tail homodimers on the surface of the virion [24]. E protein domain I (DI) is a structurally central amino-terminal domain, which is composed of a nine-strand mixed β-barrel structure. It acts as a bridge-like hinge linking extended domain II (DII) and globular domain III (DIII) via short flexible loops [31721]. DII is formed from two extended segments that project from DI. The large segment of DII contains a highly conserved loop at its tip, called a fusion peptide. The fusion peptide is responsible for the acid-catalyzed type II fusion process [1321]. DIII, a carboxyl-terminal immunoglobulin (Ig)-like structure, is located on the opposite side of DI and contains several epitopes that can induce neutralizing antibody [5].
As an enveloped virus, the first step in cell entry of Flavivirus involves binding of the E protein to a cellular receptor. Crystallographic study has revealed that the interaction between Dengue virus and dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) is mediated through carbohydrate moiety at Asn67 in DII [25]. Japanese encephalitis virus interacts with DC-SIGN through N154 in the central domain of DI [35]. Dengue virus and Japanese encephalitis virus can bind to glycosaminoglycans (GAGs) on host cells and E residues 279 to 297 are identified as involved in this binding process [22]. DIII of West Nile virus is documented to mediate virus binding to cellular receptor αVβ3 integrin [12]. Moreover, the DE loop of DIII is reported to have receptor-binding motifs participating in Japanese encephalitis virus's entry into BHK-21 cells [13]. Flavivirus may use multiple receptors for cell entry, receptors that may be strain-specific and/or cell type-dependent [20]. Many other cellular proteins have been shown to be associated with flaviviral infection, such as heat shock protein (HSP) [7], vimentin [15], and 37/67 kDa laminin [32]. However, how the E protein binds to these receptors has not been fully described, and a specific receptor is thought to be required.
HSPA9, a member of the heat shock protein 70 family, has numerous functions, such as protein folding, cytoplasmic chaperone, and peptide presentation to the immune system [27]. Recent study has shown that HSPA9 exists on the surface of DF-1 cells, and it was identified as a putative receptor for TMUV [19]. Indirect immunofluorescence assay demonstrated colocalization of HSPA9 and TMUV on the cell surface [19], but how E protein participates in the interaction with HSPA9 has not been elucidated.
In this study, we analyzed the E protein binding domains that are required for HSPA9 binding. To verify the minimal motif involved in TMUV-HSPA9 binding, peptides covering DI and DII were synthesized, and HSPA9 binding analysis was conducted. The results revealed that E protein residues 19 to 22 and 245 to 252 constitute domains that mediate TMUV binding to HSPA9.
Materials and Methods
Cells, viruses, and antibodies
DF-1 cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% inactivated fetal calf serum (FCS) in 5% CO2 at 37℃. TMUV JS804 was isolated by our laboratory and propagated in DF-1 cells as previously described [11]. All the experiments were approved by the ethical committee of Jiangsu Academy of Agricultural Sciences (approval No. SYXK-2015-0020). The titer of virus is calculated as TCID50 (50% tissue culture infective dose) according to the Reed-Muench method [26]. In brief, Serial 10-fold dilutions of the stock virus were inoculated into a monolayer of DF-1. After two-hour incubation at 37℃, inoculum was removed and cells were overlaid with a mixture of 1% agarose and cell culture media. Plaque formation was continuously observed daily for four days and TCID50 was calculated according to the method of Reed-Muench. TMUV-monoclonal antibody was generated and maintained by our laboratory as previously described [8]. Alkaline phosphatase and horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG were purchased from the Beyotime Institute of Biotechnology (China). Anti-HSPA9 (ab171089, mouse) was purchased from Abcam (UK).
Protein expression and purification
The DI (1–50 aa, 132–197 aa, 282–298 aa), DII (51–131 aa, 198–281 aa), and HSPA9 gene were synthesized by Nanjing Genscript Biotechnology (China). A synthesized gene fragment of DI or DII was subcloned into EcoR I and Sal I sites of bacterial expression vector pGEX-4t-1 (pGEX-DI or pGEX-DII). The HSPA9 gene was ligated into pET32a vector between Bam HI and Xho I sites (32a-A9). DIII was amplified by polymerase chain reaction (PCR). Primers used for cloning were: forward primer, EDIIIF, 5′-GAATTCATGGACTACAAAGACGACGACGACAAAAAGCTGAAAGGAATGACC-3′ and reverse primer, EDIIIR, 5′-GCGGCCGCTCATTTGTCGTCGTCGTCTTTGTAGTCTTGTGCTCCTTTGAGTGTT-3′. The EcoR I and Not I restriction sites are underlined and the FLAG tag (DYKDDDDK) sequences are italicized. The PCR product was ligated into pET32a vector between the EcoR I and Not I sites (32a-DIII).
Proteins were expressed in BL21 (DE3) transformed with pGEX-DI, pGEX-DII, 32a-DIII or 32a-A9. Transformed BL21 was grown in LB with shaking at 37℃. When the optical density at 600 nm (OD600) of the bacteria culture reached 0.4 to 0.6, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM for an additional 5 h. After induction by IPTG, cells were harvested by centrifugation. The recombinant proteins were purified by glutathione-agarose (Sigma, USA), anti-FLAG M2 Affinity Gel (Sigma) or High-Affinity Ni-NTA Resin (Novagen, USA) according to the manufacturer's instructions. After purification, the proteins were detected by performing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and confirmed by Western blotting. The protein concentration was detected by using a BCA kit according to the manufacturer's instructions (Beyotime Institute of Biotechnology). Proteins were then quickly frozen and stored at −80℃.
Western blot
The HSPA9-binding domains in E protein were determined by Western blotting. The purified recombinant HSPA9 protein was separated by SDS-PAGE and transferred to polyvinylidene fluoride membrane. The membrane containing the transferred proteins was blocked with 5% bovine serum albumin (BSA) in PBST (phosphate buffered saline with Tween 20; NaCl 137 mM, KCl 2.7 mM, Na2HPO4 4.3 mM, KH2PO4 1.4 mM, pH to 7.4, with 0.05% Tween 20) at 37℃ for 2 h and then incubated with purified recombinant DI, DII, or DIII protein overnight at 4℃. After being washed three times with PBST, TMUV-positive serum was added to the membrane and incubated at 37℃ for 1 h. Subsequently, the membrane was incubated with horseradish peroxidase-conjugated goat anti-mouse IgG. Finally, the signal was developed using BeyoECL Plus Kit (Beyotime Institute of Biotechnology) in accordance with the manufacturer's protocol.
Co-immunoprecipitation assay
To confirm which domain in E protein participated in HSPA9 binding, a co-immunoprecipitation assay was carried out as previously described [16]. Briefly, the purified recombinant DI, DII, or DIII protein was incubated with purified recombinant HSPA9 protein on a rocker at 4℃ overnight followed by incubation with anti-HSPA9 antibody for 5 h. Protein A/G agarose beads were added to each reaction tube and incubated on a rotator at 4℃ for 3 h. After centrifugation, the beads were collected and washed by PBS for five times. Subsequently, beads-bound immune complexes were resuspended in 4× SDS-PAGE loading buffer, boiled for 5 min, and analyzed by SDS-PAGE.
Overlapping peptide design and synthesis
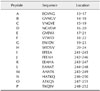
A library of 16-mer peptides, overlapping by four amino acids, was derived from DI and DII. The sequences and positions of these peptides are shown in Table 1. All peptides were synthesized and purified by Nanjing Genscript Biotechnology. The peptide purities were > 95%.
Peptide binding assay (indirect enzyme-linked immunosorbent assay [ELISA])
The HSPA9-binding peptides were detected by indirect ELISA as previously described [8]. The 96-well ELISA plate was coated with purified recombinant HSPA9 (4 µg per well) at 4℃ overnight and blocked with 1% BSA in PBST at 37℃ for 2 h. The plate was incubated with different overlapping peptides in triplicate for 1 h at 37℃. After being washed three times with PBST, the plate was incubated with TMUV-positive serum followed by incubation with HRP-conjugated goat anti-mouse IgG. Then, the plate was washed three times and subsequently incubated with TMB substrate. The reaction was stopped with 2 M H2SO4, and the OD450 nm value of each well was determined by using a BioTek (USA) microplate reader.
Truncated peptides design and synthesis
A series of truncated peptides derived from peptides 2 and 21 was designed and synthesized by Nanjing Genscript Biotechnology. The purities of the peptides were > 95%. The sequences of these peptides are shown in Table 2. Peptides A to H were derived from peptide 2 and peptides I to P were derived from peptide 21.
Dot blot analysis
Two microliters of 5 mg/mL truncated peptides were spotted on a nitrocellulose membrane. The membrane was blocked by 5% BSA at 37℃ for 2 h. Purified recombinant HSPA9 protein was added to the membrane and incubated at 4℃ overnight. Unbound HSPA9 protein was removed by washing three times with PBST. The membrane was then incubated with anti-HSPA9 antibody followed by incubation with alkaline phosphatase conjugated goat anti-mouse IgG. The signal was detected using Alkaline Phosphatase Assay Kit (Beyotime Institute of Biotechnology) in accordance with the manufacturer's protocol.
Toxicity assay
Peptide cytotoxicity was measured by using Cell Counting Kit-8 (CCK-8; Dojindo, China) according to the manufacturer's protocol. Briefly, 100 µg/mL of peptides were added to DF-1 cells in a 96-well plate and incubated for 1 h at 4℃. After being washed three times by PBS, DMEM with 10% FCS (100 µL per well) was added. Simultaneously, 10 µL CCK-8 was added per well and incubated with the cells at 37℃ for 2 h. The OD450 nm value at was measured by using a BioTek microplate reader.
Competitive receptor binding assay
DF-1 cells were incubated with 50 µg/mL of peptides or BSA (as control) at 4℃ for 1 h. Unbound peptides were removed by washing the cells twice with chilled PBS. The cells were then infected with 200 TCID50 TMUV for 30 min at 4℃ followed by 1 h at 37℃. Unbound virus was removed by washing twice with PBS. Virus RNA was extracted by using a QIAamp Viral RNA Mini Kit (QIAGEN, Germany), and TMUV RNA levels were determined by quantitative reverse transcriptase (qRT)-PCR as described previously [39]. The primers used for real-time RT-PCR were: EF primer (forward, 5′-GTGAGATCTTACTGCTATGAG-3′) and the ER primer (reverse, 5′-ACTTGGCACATGTCTGTATGC-3′). Real-time RT-PCR data were analyzed using the comparative cycle threshold (CT) method (ΔΔCT). Beta-actin was chosen as a reference gene for internal control. Differences between the CT values of the target gene and the internal control (ΔCT = CTtarget − CTinternal control) were calculated to normalize the differences in the amount of total complementary DNA added to each reaction and the efficiency of the real-time RT-PCR. The negative control was used as a reference for each comparison. Differences between the ΔCT of each sample and the reference sample (ΔΔCT = [CTtarget − CTinternal control] sample − [CTtarget − CTinternal control] negative control) were then calculated. Three independent experiments were carried out.
Results
DI and DII mediate TMUV–HSPA9 binding
To test which E protein domain participates in the HSPA9 binding process, we performed Western blotting using recombinant HSPA9. Purified recombinant DI, DII, or DIII protein was incubated with the membrane containing HSPA9. Domains bound to HSPA9 were detected by the TMUV-positive serum. As shown in panel A in Fig. 1, a distinct band with an approximate 90 kDa molecular mass was observed in the DI and DII incubated membranes, while no band was visualized in the DIII incubated membrane, suggesting that DI and DII were able to bind to HSPA9. In order to validate the binding between DI or DII and HSPA9, co-immunoprecipitation assays were carried out. As shown in panel B in Fig. 1, DI or DII was co-immunoprecipitated with HSPA9 by the anti-HSPA9 antibody. Consistent with the Western blotting results, the band of DIII was not observed in the co-immunoprecipitated mixture containing DIII and HSPA9, confirming that only DI and DII were involved in TMUV–HSPA9 binding.
Identification of HSPA9 binding peptides derived from DI and DII
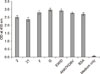
The 16-mer overlapping peptides derived from DI and DII were synthesized and analyzed for their interaction with HSPA9 by using indirect ELISA. Fig. 2 showed that two (i.e., peptides 2 and 21) of the overlapping peptides interacted with HSPA9 and exhibited the highest binding indices. Other peptides did not show the capacity to bind to HSPA9. Removing the overlapping sequence, the core sequences of peptides 2 and 21, which were involved in HSPA9 binding, were GVEWIDVV and AHATKQSV, respectively.
Location of the minimal HSPA9-binding motif by truncated peptides
Truncated peptides were synthesized according to core sequence of peptides 2 (peptides A to H) and 21 (peptides I to P). Dot blots were used to test whether truncated peptides could bind to HSPA9. The results demonstrated that peptides F and G derived from peptide 2 exhibited positive reactivity to HSPA9 (panel A in Fig. 3). The sequence shared by peptides F and G was EWID (E residues 19 to 22), which is located within loop B0 of E protein. However, the truncated protein derived from peptide 21 showed negative reactivity to HSPA9, suggesting that reduction of residues of AHATKQSV can lead to failure in HSPA9 binding. In order to validate further the minimal HSPA9-binding motif, peptides EWID and AHATKQSV were synthesized and analyzed. The Dot blot results showed that EWID and AHATKQSV bind to HSAP9 (panel B in Fig. 3). Therefore, it is concluded that EWID and AHATKQSV are required for TMUV–HSPA9 binding.
Toxicity assay
It is possible that peptides can induce cellular toxicity, which can block TMUV binding. To address this possibility, DF-1 cells were exposed to peptides at a 100 µg/mL concentration. Cell viability was determined by using CCK-8. There was no statistical difference in viable cell numbers between control cells and cells exposed to peptides (Fig. 4), indicating that the peptides are non-toxic to DF-1 cells.
Competitive inhibition of TMUV binding by peptides
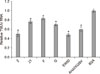
To test whether peptides binding to HSPA9 can compete with endogenous E in mediating virus binding, we performed virus binding assays and determined the viral RNA level by using qRT-PCR. As shown in Fig. 5, the viral RNA level was significantly lower in peptide 2-, 21-, F-, G-, EWID-, and AHATKQSV-treated DF-1 cells compared with that in BSA-treated cells, indicating that virus binding to DF-1 cells was competitively inhibited by peptides 2, 21, F, G, EWID and AHATKQSV.
Discussion
The first step in the Flavivirus infection process involves binding of E protein to a cellular receptor. This interaction initiates a chain of dynamic events that enable viral entry into the cell [27]. Flaviviruses can recognize ubiquitous cellular molecules or utilize multiple receptors for cell binding as flaviviral infection has been observed in different cell types [28]. The initial binding between Flavivirus and a cellular receptor on the surface of sensitive cells often determines tropism and pathogenesis of the virus [12].
Numerous studies have suggested that the infection spectrum of TMUV is wide, and it has been detected duck, goose, chicken, pigeon, sparrow, and mouse. Tembusu RNA can also be detected in duck industry workers [31]. The results of in vitro culture show that TMUV replicates well in cells of different origins, including BHK-21, Vero, Hela, DEF, GEF, DF-1 and C6/36 cells [293437]. Since both E protein and the host components involved in virus entry were reported to be primary determinants of host range, cell tropism, and virulence, further investigation of the TMUV E protein-associated receptor-binding motif may provide novel insights into the transmission and evolution of TMUV.
In this study, we focused on the HSPA9 binding motif of E protein. Western blotting and co-immunoprecipitation assays identified DI and DII as the major binding domains of the virus to cellular receptor HSPA9. Overlapping peptide binding assays demonstrated that GVEWIDVV and AHATKQSV were involved in HSPA9 binding. Next, the minimal binding motif was identified by Dot blots using truncated peptides derived from GVEWIDVV or AHATKQSV. The results showed that EWID was required for TMUV–HSPA9 binding. Surprisingly, truncated peptides from AHATKQSV exhibited negative reactivity to HSPA9. It was speculated that all residues of AHATKQSV were indispensable for HSPA9 binding. In order to uphold this speculation, EWID or AHATKQSV were synthesized and used in Dot blot assays. The Dot blot results demonstrated that EWID and AHATKQSV bound to HSPA9. Competitive binding assays showed that pre-incubation of EWID or AHATKQSV can effectively inhibit TMUV infection, indicating that both motifs were necessary for TMUV–HSPA9 binding.
The motif AHATKQSV contains an ij loop (AHATKQ) according to the nomenclature used for class II proteins. The ij loop, connecting β-strands i and j, is located at the tip of the second segment of DII and is presumed to play a critical role in Flavivirus membrane fusion. A virus that contains mutations in the ij loop is completely noninfectious and not active in membrane fusion [4]. Sequence alignments of the ij loop indicate that histidine (H246) is located at the same position and is completely conserved among all Flavivirus spp. [38]. That highly conserved histidine has an important role in the regulation of E protein oligomerization and promotion of viral entry [923]. Substitution of H246 with alanine or glutamine impairs viral assembly and results in a significant reduction in the number of virus particles released from transfected cells [23]. TMUV is a newly emerging Flavivirus spp. that has spread widely in China and the functional determinant of its E protein is still unclear; therefore, it is intriguing and necessary to explore further the involvements of the ij loop and H246 in the TMUV infection process.
In this study, we successfully performed the first identification of the minimal motif required for TMUV binding to HSPA9 on DF-1 cells. Our results provide important information useful in deciphering the TMUV infection mechanism as well as in providing guidance for preventing and eradicating TMUV-related diseases.




 XML Download
XML Download