

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited renal disease and is characterized by numerous cysts in both kidneys1). In addition to affecting the kidneys, ADPKD is associated with various extrarenal vascular abnormalities including cerebral aneurysms, coronary artery aneurysms and dissection, cervicocephalic artery dissection, and aortic aneurysms and dissection23). Visceral artery aneurysms are rare; although gastroduodenal artery pseudoaneurysm and splenic artery aneurysm have been reported, no previous case of an esophageal artery pseudoaneurysm has been reported4).
Takayasu arteritis is a chronic granulomatous, large-vessel vasculitis primarily involving the aorta, its main branches, and the pulmonary arteries. Most arterial lesions in Takayasu arteritis are stenotic, whereas aneurysms can be found at various sites, including the visceral abdominal arteries56).
We present the case of an ADPKD patient who was diagnosed with an esophageal artery pseudoaneurysm and Takayasu arteritis.
Case Report
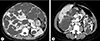
A 47-year-old female visited the emergency room due to sudden pain in her upper abdomen and back. The patient had been diagnosed with ADPKD and severe polycystic liver disease 13 years prior (Fig. 1) and had received follow-up care with no symptoms. Additionally, as a complication of ADPKD, an arachnoid cyst in the left middle cranial fossa was found on brain magnetic resonance imaging.
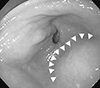
The patient's blood pressure at the time of hospitalization was 140/100mm Hg, her heart rate was 62 beats per minute, her respiratory rate was 18 breaths per minute, and her temperature was 36.3℃. The laboratory findings showed a white blood cell count of 12,770/µL, a hemoglobin count of 13.5 g/dL, a platelet count of 184,000/µL, a blood urea nitrogen level of 15mg/dL, a creatinine level of 0.85 mg/dL, and a C-reactive protein level of 0.35mg/dL. Esophagogastroduodenoscopy showed external compression in the lower gastric body, but no hemorrhage of the gastroesophageal mucosa (Fig. 2).
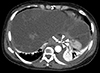
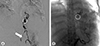
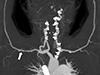
Computed tomography (CT) angiography showed an esophageal artery pseudoaneurysm and a hematoma in the esophagus (Fig. 3). Urgent angiography and embolization of the esophageal artery pseudoaneurysm were performed (Fig. 4). CT angiography was performed due to a blood pressure difference of 30mmHg (right arm, 124/77mmHg; left arm, 156/74mmHg) and the presence of a left subclavian bruit. This examination revealed that the patient also had Takayasu arteritis (Fig. 5). Autoantibody tests, including antineutrophilic cytoplasmic antibody, anticyclic citrullinated peptide antibody, and rheumatoid factor, were all negative. Positron emission tomography was performed to assess inflammation, but did not show any concerning signs of active vasculitis. The patient was transferred to outpatient management with low-dose aspirin (100mg once per day), and has not shown any signs of complications.
Discussion
Both ADPKD and Takayasu arteritis can cause vascular complications, but involvement of the visceral arteries is uncommon47). There have been no reports of the simultaneous diagnosis of ADPKD and Takayasu arteritis, and this is also the first reported case of esophageal artery pseudoaneurysm in a patient with both ADPKD and Takayasu arteritis.
ADPKD is a hereditary disorder, and is accompanied by various systemic manifestations18). Among these, vascular abnormalities, such as cerebral or coronary aneurysm and aortic aneurysm and dissection, are relatively well-known, although the mechanisms are not entirely understood2). The possible mechanisms that cause vascular abnormalities in ADPKD patients include damaged vascular integrity due to abnormal polycystin expression caused by PKD mutations and connective tissue abnormalities.
ADPKD is known to be caused by mutations in the PKD1 and PKD2 genes, which code for the polycystin 1 and polycystin 2 proteins, respectively9). These mutations cause the growth of numerous renal and extrarenal cysts in ADPKD patients8). Studies have suggested that the vascular abnormalities found in ADPKD have to do with the polycystin 1 and polycystin 2 present in the vascular smooth muscles and endothelium and their involvement in the mechanism of vascular remodeling and early aneurysm development2310).
Researchers have long suspected that ADPKD is a type of connective tissue disorder. The histopathological findings of ADPKD patients are generally suggestive of connective tissue disorders. Based on vessel aneurysmal changes in the renal tissue, as well as excessive collagen matrix and edema seen in an ADPKD patient who underwent nephrectomy, it was suggested that weak, excessive collagen is the common pathogenic factor that is responsible for various ADPKD symptoms11). Moreover, in an autopsy of an individual with vertebrobasilar dolichoectasia, changes were observed in the integrity of the extracellular matrix, with degeneration and multiple gaps in the internal elastic lamina, thinning of the media, and smooth muscle atrophy; these observations were considered to provide grounds for seeing ADPKD as a connective tissue disorder12).
Takayasu arteritis is a systemic inflammatory vasculitis of unknown etiology. The pathogenesis of Takayasu arteritis is poorly understood, but T cell-mediated mechanisms are thought to be the most important13). The inflammatory process within the vessel can result in stenosis, occlusion, dilatation, or aneurysm formation in the involved portions of the arteries5). Although many studies have investigated Takayasu arteritis, its diagnosis is still challenging. This patient fulfilled 3 of 6 American College of Rheumatology criteria for the diagnosis of Takayasu arteritis14): difference in blood pressure between arms, the presence of a left subclavian bruit, and arteriographic abnormalities. However, to meet the modified Ishikawa diagnostic criteria, esophageal artery pseudoaneurysm should be included5).
The differential diagnosis of esophageal artery pseudoaneurysm includes atherosclerosis, connective tissue disorders (e.g., Marfan, Ehlers-Danlos, or fibromuscular dysplasia), vasculitis (e.g., polyarteritis nodosa or Takayasu arteritis), and polycystic kidney disease15). In this case, the esophageal artery pseudoaneurysm may have been caused by either ADPKD or Takayasu arteritis. We reviewed the patient's images before the onset of symptoms and found celiac axis stenosis, which can be a manifestation of Takayasu arteritis.
Since this was the first report of ADPKD and Takayasu arteritis in a single patient, it was not easy to identify a possible link between the 2 diseases. Baek et al.16) proposed the possibility of a T cell-mediated autoimmune response to an abnormal extracellular matrix protein resulting from the mutation implicated in Marfan syndrome, with regard to the pathogenesis of a case of Marfan syndrome accompanied by Takayasu arteritis. In this report, we suggest that Takayasu arteritis may be caused by a T cell-mediated autoimmune response to abnormal polycystin proteins, similar to the proposal by Baek et al.
We presented a novel and interesting case of esophageal artery pseudoaneurysm in a patient with ADPKD, proving that ADPKD is a type of connective tissue disorder. Vascular abnormalities are a major complication of ADPKD, but if rare vascular complications occur in ADPKD patients, it is important to reassess the possibility of other forms of vasculitis or another connective tissue disorder. Further study is required to confirm the relevant mechanisms.
 XML Download
XML Download