

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Recently, there has been growing interest in the relationship between cardiac diseases and diet. A long-term, high-salt (NaCl) diet leads to chronic hypernatremia and overall cardiac hypertrophy1). Left ventricular hypertrophy increases the possibility of arrhythmia and even sudden cardiac death2). In addition to a high-salt diet, angiotensin II (Ang II) is a well-known factor that induces cardiac hypertrophy through the renin-angiotensin system(RAS), and it has been used to produce models for cardiac hypertrophy34). However, the direct cellular effects of a high-NaCl diet are not yet clearly understood.
In cardiac hypertrophy, upregulation of early gene expression occurs, such as the expression of sHSP5). Moreover, late embryogenesis abundant (LEA) proteins have similar roles in development, and they are classified into groups, according to their sequence and function67). LEA proteins in group 3 (LEA3), found in Artemia, play protective roles during hypersalinity8). sHSP and LEA3 have similar protective functions, when an organism faces hyperosmolality and various stresses6). However, there is no previous study that shows the relationship between sHSP and LEA proteins.
HSP22 (heat shock protein 22, HspB8, and H11 kinase) is a sHSP that is expressed abundantly in the heart. HSP22 is a mediator of cardiac hypertrophy, and it plays a key role in the regulation of apoptosis, development, and intracellular Ca2+ ions91011121314). The role of HSP22 in cardiac hypertrophy in chronic hypernatremia is currently unknown. Therefore, the aim of the present study was to establish an in vitro hypernatremic model with cardiac cellular hypertrophy, to demonstrate the effectiveness of this model by comparing 2 hypertrophic factors (high NaCl and Ang II), and to identify the LEA like-properties of HSP22 in these 2 different cellular models.
Methods
1. Cell Culture
The rat cardiomyoblast-derived H9C2 cell line was purchased from the Korean Cell Line Bank. The H9C2 cells were maintained in Dulbecco's modified Eagle's medium supplemented with fetal bovine serum(FBS; 10%), penicillin (100 µg/mL), and streptomycin (100 µg/mL) (DMEM) in 5% CO2 humidified air at 37℃.
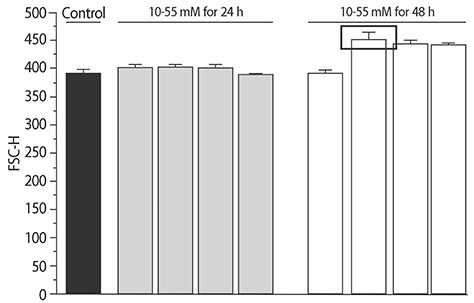
2. In vitro hypernatremic model
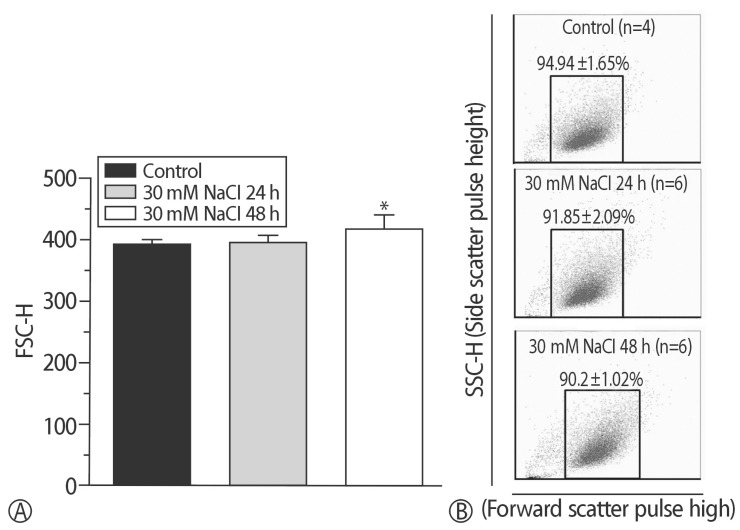
The added concentration and maintenance time for NaCl was selected using the forward side scatter pulse height (FSC-H) values from flow cytometry (Table 1, Supplement 1). Based on this, 30mM NaCl was added to the DMEM. The media were filtered through 0.4 µm filters. Cells were maintained for 48 h to produce the in vitro hypertrophic models.
3. Flow cytometric analysis
Suspended H9C2 cells in PBS (phosphate-buffered saline) were used. To exclude osmotic effects, the cells were incubated in normal osmotic PBS for 30min. After incubation, 10,000 cells were automatically analyzed by FSCH to measure the cell size and side scatter pulse height (SSC-H) in order to study the intracellular granules (FACS Calibur™).
4. Confocal analysis
Cells were fixed in −20℃ methanol for 5 min at room temperature (RT). The cells were washed 3 times with ice-cold PBS and then blocked with 10% FBS in PBS for 30 min at RT. After blocking, the cell nuclei were stained with 1 µg/mL of 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) in 3% FBS and PBS for 30 min at RT. After staining with DAPI, the images were visualized with a confocal microscope and a coupled image analysis system(Nikon).
6. Cell viability
The trypan blue exclusion test was performed17). The results were calculated using the following equation: (the total number of viable cells/the total number of cells)×100.
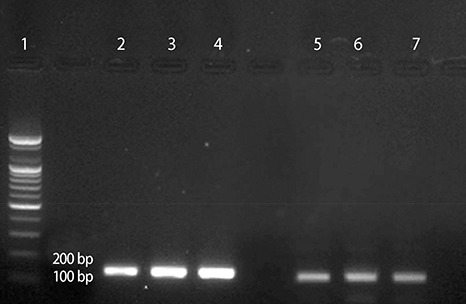
7. Real-time reverse transcriptase PCR(RT-PCR) and DNA gel electrophoresis
Total RNA was extracted. The RNA quantity and quality were measured using the 260/280 ratio obtained from a Nano-drop spectrophotometer. Using RT-PCR, cDNA was synthesized from RNA. For the analysis of mRNA expression, forward and reverse primers were used (Table 2). One µL of each set of primers was used with TOPreal™ qPCR premix (10 µL), nuclease-free water (20 µL), and a cDNA sample (1 µL). The Tm value of the primers was 58℃. After an initial 10 min at 95℃, 45 cycles of the following protocol were completed: 95℃ for 10s, 60℃ for 15s, and 72℃ for 30s. The fold changes of HSP22 were compared with those of a house keeping gene (18s ribosomal RNA(rRNA)). The data were analyzed using the ΔCt method18). To confirm the primer activities, an ethidium bromide-stained agarose gel (2%) was used for DNA gel electrophoresis. The size of the HSP22 product was 129 base pairs (bp), while that of the 18s rRNA product was 135 bp.
8. Intracellular Ca2+
The cells were loaded with 2mM of the fluorescence probe Calcium Green-1™ in DMEM for 2 h. Cells were washed twice with PBS, and 10,000 cells were analyzed using flow cytometry. Single cells were observed under a confocal microscope after fixation with ice-cold methanol.
9. Statistics
All data were expressed as the mean±S.E., and n indicated the number of cells used. For all tests, cells were obtained from a minimum of 3 different culture dishes for each treatment and experiment. Analysis of the data was conducted using Student's t-test and one-way ANOVA. p<0.05 was considered to be significantly different in the statistical analyses.
Results
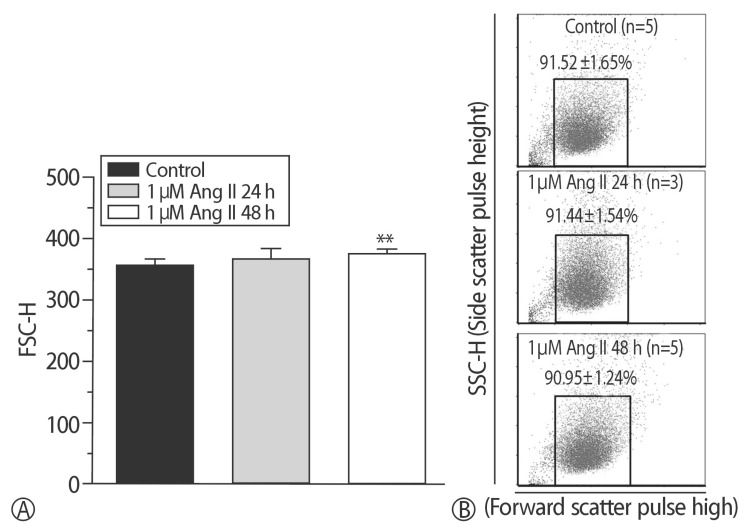
1. Supplementation with 30mM NaCl and 1 µM Ang II for 48 h affects the size of cells without inducing abnormal cytological changes
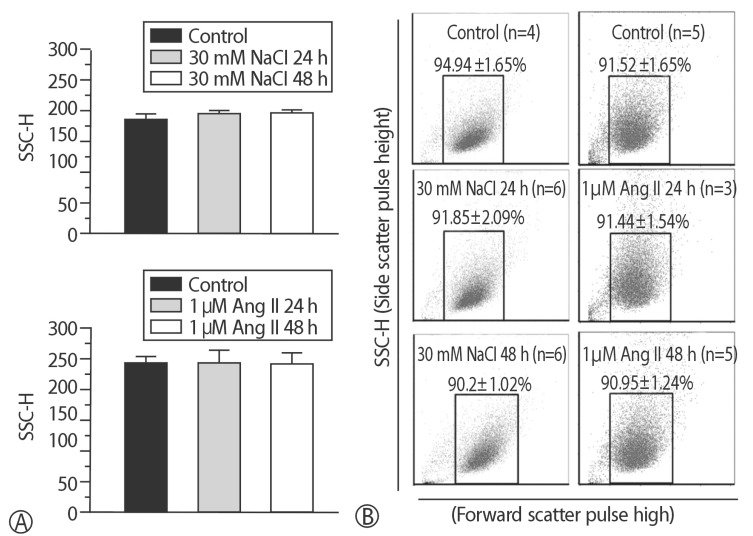
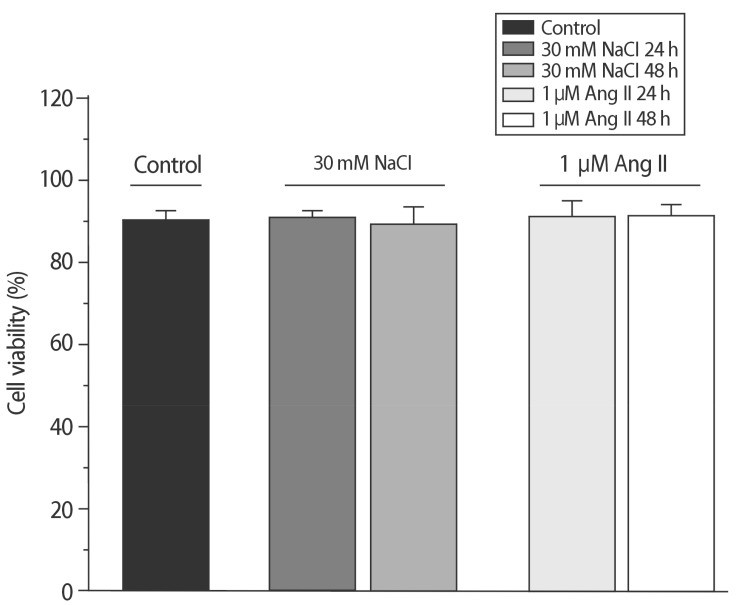
To compare the characteristics of the in vitro hypernatremic model, an Ang II-induced hypertrophic model (Ang II model) was used, because treatment with Ang II induces cellular hypertrophy in H9C2 cells, which mimics the hypertrophic responses of primary cardiomyocytes3). First, the FSC-H values significantly increased following supplementation with 30 mM NaCl (p<0.05) and 1 µM Ang II for 48 h (p<0.01) (Figs. 1 and 2), indicating an increase in the size of the cells. The mean of the FSC-H was 391.21±6.63 for the control group. The mean of the FSC-H for the 24 h group with 30mM NaCl did not change. However, the mean of the FSC-H for 48 h was 416.49±17.08. The mean FSC-H value was 356.77±9.26 for the control group. The mean of the FSC-H for the 24 h group with 1 µM Ang II did not exhibit a statistically significant change. However, the mean of the FSCH value for the cells maintained for 48 h was 375.09±7.61. There were several factors that could have affected the control samples. For this reason, different controls were used in each experiment to ensure an accurate comparison. The difference in values was more evident for the supplementation with NaCl. Second, SSC-H was constant among the controls after treatment for 24 h and 48 h with each hypertrophy-inducing agent (Fig. 3). Third, the nuclear and cell morphologies were similar among the controls and the 2 models under confocal microscopy with DAPI staining (Supplement 2). Our hypertrophic model, induced by the addition of NaCl, contained less granular content in the cytoplasm than other models. The results indicated that the addition of 30mM NaCl did not strongly affect the cells. Finally, the cell viability was examined (Fig. 4). For all of the groups, there were no changes in the cell viability. Therefore, this model did not exhibit abnormal morphological changes, and the cellular hypertrophy induced by these conditions was termed the in vitro hypernatremic model.
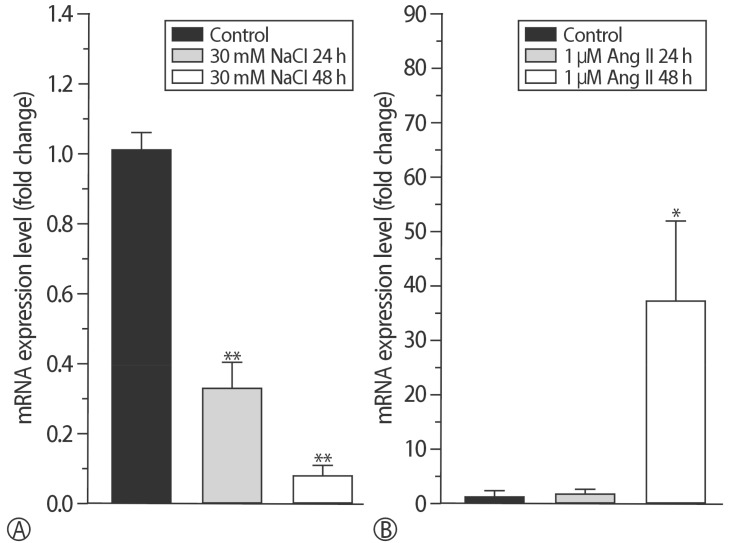
2. Different mRNA expression patterns of HSP22 between the 2 different cellular models
RT-PCR was conducted to compare the mRNA expression patterns after exposure to Ang II or NaCl for 24 or 48 h. HSP22 was compared with a house-keeping gene. The mRNA expression of HSP22 did not increase after treatment with Ang II for 24 h, but the expression of HSP22 increased after 48 h. In contrast to the Ang II model, the expression of HSP22 in the in vitro hypernatremic model gradually decreased in a time-dependent manner (Fig. 5). After these experiments, DNA gel electrophoresis was used to confirm the product of each primer set used for RT-PCR (Supplement 3). The results indicated that the design of the primers and PCR analysis were conducted appropriately.
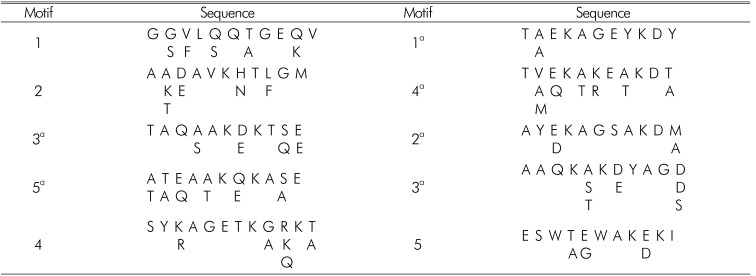
3. Amino acid sequence homology between HSP22 and LEA3
A gradual decreasing pattern, in response to high NaCl, has been shown for LEA3 in Artemia19). Based on the similarity of the decreasing patterns for LEA3 in Artemia and HSP22, a comparison of the amino acid sequences was conducted. LEA3 proteins have several types of conserved domains (Table 3)20). The sequences of these conserved domains were also compared. The consensus sequence of HSP22 was found at residues 108-128, which corresponds to residues 117-137 of LEA3 from Artemia. The identities were 33%(7/21); 57%(12/21) were positive, there were 0 gaps, and the expectation was 3.9. The similarity was meaningful, because part of the α-crystalline domain of HSP22 was similar to 3a motif sequence of LEA3. The similar expression patterns and amino acid sequences for HSP22 and LEA3 indicated that HSP22 might have an LEA-like function20).
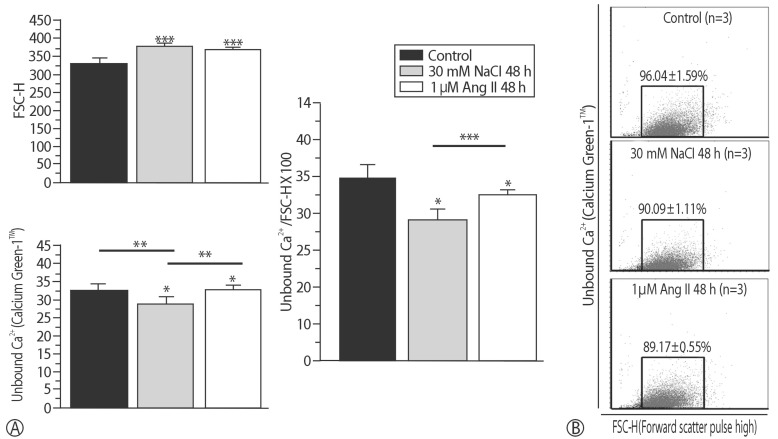
4. HSP22 affects the intracellular homeostasis of free Ca2+
To determine the roles of HSP22 in hypertrophy, the intracellular Ca2+ level was compared using a Ca2+-binding dye for the control; the FSC-H increased for the 2 hypertrophic models (p<0.001). Unbound Ca2+ increased in the Ang II model (p<0.05). However, compared with the Ang II model, Ca2+ significantly decreased in the in vitro hypernatremic model (p<0.01). To compensate for the size of the cells using the equation: unbound Ca2+/FSC-H×100, the corrected Ca2+ decreased in the Ang II model (p<0.05), and the decrease in Ca2+ was more evident with NaCl (p<0.001) (Fig. 6). The corrected result showed that size of the 2 hypernatremic models increased as the intracellular free Ca2+ decreased. Moreover, Ca2+ was confirmed in a single cell (Fig. 7). The results indicated that the role of HSP22 in cellular hypertrophy is related to impaired intracellular Ca2+ homeostasis.
Discussion
Hypertrophy in cardiomyocytes is a key factor in the pathogenesis of cardiac diseases, such as heart failure, arrhythmia, and sudden cardiac death21). Many previous studies have demonstrated the causes of hypertrophy, including changes in metabolism, RAS, and Ca2+-dependent transcription factors222324252627). However, the mechanism of cellular hypertrophy, induced by a chronic high-NaCl diet, has not been elucidated.
Recently, the re-induction of cardiac embryonic genes, through a process called the “immediate early gene program,” has been reported in several studies using cardiac disease models5282930). sHSPs are cardiac embryonic genes that play protective roles in the heart3132). LEA proteins are also known to be protectors and regulators of cellular development in various organisms620). In particular, the LEA3 proteins protect the cell during changes in osmolality8). Using the BLAST program, similar sequences that protect against osmotic damage were found in both HSP22 and LEA3 from Artemia. The α-crystalline domain is an important part of the sequence of HSP2233). The homology identified in the α-crystalline domain was term ed “LEA-like-HSP22.” All of these data imply that proteins that regulate development, such as sHSPs and LEA, play key roles in the process of hypertrophy under conditions of high osmolality. Specifically, LEA-like-HSP22 has diverse functions in the heart91011121334).
Having been incubated in normal osmotic PBS for 30 min, an increase in FSC-H was sufficient to indicate that hypertrophy had been induced in our in vitro hypernatremic model. The lack of abnormal changes in SSC-H, viability, and morphology across the in vitro hypernatremic model, control, and Ang II model shows that our model is not negatively affected by the addition of NaCl. In a previous study, an increase in SSC-H was observed in damaged cells35). Our hypernatremic conditions could more effectively increase the size of cells than the conditions obtained with Ang II, because the increase in FSC-H in our model was greater than that observed for the Ang II model. Although excitation-contraction coupling (E-C coupling) by action potentials is not found in H9C2 cells, previous studies have shown that the intracellular Ca2+ can be changed and that Ca2+ can be a mediator of cellular hypertrophy in H9C2 cells36373839404142). Therefore, the intracellular Ca2+ homeostasis is important in H9C2 cells.
With our model, our group anticipated that HSP22 would increase to protect the cells in the high-NaCl environment. Contrary to our expectations, HSP22 decreased in our model, and this trend was not observed with the Ang II model. This result suggests that HSP22 may participate in the protective process of cardiac hypertrophy with chronic hypernatremia and the protective process of intracellular Ca2+ homeostasis. This agrees with our finding that the Ca2+ in the hypertrophic H9C2 cells changed with the different HSP22 expression levels, because Ca2+ is used as a second messenger in the heart, and Ca2+ has important functions, including the regulation of metabolism, E-C coupling, and hypertrophic processes43444546). Our results indicate that HSP22 participates in the intracellular Ca2+ homeostasis, because in our model, the impaired, intracellular free Ca2+ homeostasis and an increased cell size were more evident with the decreased HSP22 expression than other for the groups.
In summary, the expression of HSP22 was significantly different in the 2 hypertrophic models, and it is important for the maintenance of the intracellular Ca2+ homeostasis in H9C2 cells. However, the present study had several limitations, including the properties of HSP22 in contractible cardiomyocytes and the direct relationship between cardiac hypertrophy, HSP22, and Ca2+ in the hypertrophic process. These limitations need to be explored.
Conclusion
We conclude that a high-NaCl condition induces cardiac cellular hypertrophy, due to impaired intracellular Ca2+ homeostasis. It is related to changes in the LEA-like HSP22 expression pattern. This observation can be used for the development of new therapies that can treat cardiac hypertrophy, induced by a chronic high-NaCl diet. However, the protection of the intracellular Ca2+ homeostasis with a regulated LEA-like HSP22 expression pattern must be confirmed in vivo and in patients.




 XML Download
XML Download