

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Cerebral amyloid angiopathy (CAA) involves cerebrovascular pathology, which is caused by the progressive accumulation of amyloid-beta (Aβ) in the media and adventitia of the cortical and leptomeningeal vessels.12 Previous reports have shown that, while approximately 90% of patients with Alzheimer's disease (AD) exhibit CAA pathology, only 25% have severe CAA pathology.3
With recent advances in AD pathobiology, clinical trials of amyloid-targeting therapies have been widely conducted. However, increasing evidence suggests a connection between amyloid-targeting agents and amyloid-related imaging abnormalities (ARIA), including vasogenic edema and hemorrhage.4 Microbleeds (MBs) are characteristic of ARIA hemorrhages. While the pathophysiology of ARIA is still unknown, patients with preexisting MBs are vulnerable. Furthermore, severe CAA in impaired vasculature may be implicated in ARIA development.5 Detecting MBs in AD patients has therefore generated increasing interest to prevent causing side effects in immunotherapy trials. We report a patient with earlyonset AD (EOAD) who, despite not having CAA-relating markers on MRI, exhibited CAA pathologies at autopsy.
CASE REPORT
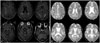
A 62-year-old ambidextrous man visited our memory clinic. His memory began to decline at the age of 59 and gradually deteriorated thereafter. By the age of 61, his family had begun to notice that he had developed visuospatial dysfunction, personality changes, and abnormal behaviors. He further demonstrated impaired activities of daily living, such as carelessness concerning personal hygiene, poor judgement in urgent situations, and difficulty in the work place. He was an accountant with 18 years of education. No family history of dementia or other neurologic diseases were reported. Upon neurological examination, he showed rigidity in the axial and right upper limb. Dressing apraxia, dyscalculia, and right-left disorientation were also observed. No other abnormal neurologic signs were evident. His Mini-Mental State Examination (MMSE) score was 19. With the exception of attention, detailed neuropsychological tests further revealed marked impairment in most domains. His apolipoprotein E (APOE) genotype was ε4/ε4. An axial fluid-attenuated inversion recovery (FLAIR) image on 3T brain magnetic resonance imaging (MRI) showed mild atrophy in the bilateral medial temporal and frontal lobes. Mild white matter hyperintensities (WMHs), rated according to the modified Fazekas scale, were found in FLAIR images (Fig. 1A).6 Gradient-echo T2* (GRE) images showed no abnormalities (Fig. 1B). These findings fulfilled the Neurological and Communicative Disorders and Stroke-Alzheimer's Disease and Related Disorders Association (NINCDS-ADRDA) criteria for probable AD.7 After the diagnosis was determined, the patient was prescribed an acetylcholinesterase inhibitor. His symptoms remained stable for the following 5 months, but declined sharply to a MMSE score of 3 at the age of 63. Although FLAIR images demonstrated more atrophy and moderate WMH than that observed in previous imaging, axial GRE images showed no signal loss. Pittsburgh compound B (PiB) positron emission tomography was positive, and PiB uptake was found in the bilateral frontal, lateral temporal, and parietal lobes. The patient was admitted to a hospital with pneumonia and died of aggravated pneumonia at 67 years. A postmortem autopsy was performed.
The postmortem examination revealed a decreased brain weight of 985 g (standard for age: 1360 g). The bilateral frontal, parietal, and temporal lobes featured significant atrophy. Hippocampal atrophy, pallor of the locus coeruleus and substantia nigra, and moderate ventricular dilatation were also observed. The thalamus, basal ganglia, brain stem, and cerebellum were relatively well preserved. Pathologic examination was performed on 13 sections using routine hematoxylin & eosin (H&E) (Fig. 2). Luxol fast blue, Bielschowsky silver stain, and immunohistochemical (IHC) staining for Aβ (Biolegend; 4G8), phosphorylated tau (Thermo scientific; AT8), TAR DNA-binding Protein-43 (TDP-43) (Cosmo bio; polyclonal), neurofilament (Dako, 2F11), GFAP (Novocastra, GA5), and α-synuclein (Abcam; polyclonal) were performed according to the National Institute on Aging-Alzheimer's Association (NIA-AA) guidelines for AD neuropathologic changes (ADNC).
The neuropathologic findings corresponded to A3 (Thal phase 5), B3 (Braak and Braak stage VI), and C3 (Consortium to Establish a Registry for Alzheimer's Disease; CERAD score frequent), indicating a high level of ADNC (Fig. 3A–C). No evidence of TDP-43 expression or Lewy body pathology was seen. Additionally, amyloid angiopathy was identified throughout cerebral, cerebellar, and mid brain parenchyma, as well as in almost all leptomeningeal vessels in the cerebral cortex, cerebellum, and brain stem. These observations were rated as severe according to the scales by Olichney, et al.8 and Vonsattel, et al.9 Aβ IHC and H&E staining revealed amyloid angiopathy (Fig. 3D and E). Perivascular microhemorrhages were associated with parenchymal amyloid angiopathy. The final diagnosis was AD with amyloid angiopathy.
This patient provided written informed consent and the study was approved by the Institutional Review Board of Samsung Medical Center (2016-11-032-005).
DISCUSSION
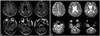
This report describes a case of EOAD with severe CAA burden. The patient was an APOE ε4 homozygote, which rendered him prone to CAA pathology.10 However, the last MRI (Fig. 4), taken at 64 years of age and 28 months before death, did not reveal any CAA-related markers.
Several pathologies are related to CAA, including microatheroma, microaneurysm, and microhemorrhage. MBs detected by GRE images represent microhemorrhages that result from the collapse of a microaneurysm. Though the patient's brain exhibited a severe CAA burden, MRI did not reveal any MB. Parameters of the MRI may account for this discrepancy. Thinsection susceptibility-weighted imaging (SWI) and a higher field strength may have detected cerebral microbleeds (CMB), a CAA-related marker, where the GRE proved insufficient.11 Another possible explanation may be underestimated CMB by MRI with respect to histopathology.12
Our report has two limitations. Although we used the latest MRI, there was a 28-month gap between MRI and autopsy. Therefore, the GRE could not show the CMB and exact CAA burden at the time of death. Second, as aforementioned, SWI is more sensitive than GRE and might have detected CMB.
In conclusion, although CMB on MRI may indicate microhemorrhage in advanced CAA, CMB might not appear on brain imaging. CAA should be considered as a combined pathology in AD patients who are APOE ε4 homozygotes, even if CMB does not appear on MRI.


 XML Download
XML Download