

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Unverricht-Lundborg disease (ULD) (EPM1, OMIM 254800) is the “purest” type and the most common form of progressive myoclonic epilepsy (PME).12 It is characterized by symptoms of myoclonic jerks and generalized tonic-clonic seizures (GTCSs), which are caused by photic or touching stimulus and usually occur at the age 6–16 years.3 This disease is an autosomal recessive disorder, and the CSTB gene encoding cystatin B, a cysteine protease inhibitor, is the only gene known to be associated with ULD.4 The 12-nucleotide dodecamer repeat (5′-CCC-CGC-CCC-GCG-3′) expansion in the promotor region of CSTB accounts for approximately 90% of cases worldwide and 99% of the EPM1 cases in Finland.5 Loss of lysosomal linkage and an endogenous neuroprotective role of CSTB are known as the main pathological mechanisms of the disease.6 Here, we report a patient with ULD and her families diagnosed by Southern blot analysis. It is the first case of a genetically confirmed ULD in Korea. This study was approved by the Institutional Review Board of Severance Hospital.
CASE REPORT
A 20-year-old woman presented with both uncontrolled hand tremor and seizure attacks. The perinatal and infancy periods of the patient were not eventful, and early developmental milestones were normal. She developed myoclonic movements of the whole body at the age of 10 years. The myoclonic movements became aggravated gradually with more severe involvement of both upper limbs. They occurred frequently every day, more severe on settling into sleep and awakening, and their severity was reduced after turning off the light. They were provoked upon sudden movements, such as standing up. Also, tremulous movements of both hands (small amplitude jerks) occurred persistently. They increased in severity as the hand moved closer to its target; therefore, detailed hand movements, such as when operating a cell phone, were difficult. Also, she could not run because of provoked myoclonic movements since the age of 13 years. Dysarthric speech and gait disturbance were recognized around that time and became gradually aggravated. She had developed mild intellectual disability since the age of 10 years. The results of neuropsychological tests (Korean-Wechsler Intelligence Scale for Children-III) taken at the age of 15 years showed mild mental retardation [full-scale intelligence quotient (IQ)=65, verbal IQ=61, and performance IQ=75]. Also, she showed emotional liability. GTCSs following a series of myoclonic movements occurred for the first time, a half year after the occurrence of myoclonus. GTCSs were mainly provoked by bright lights or emotional stress and occurred once a week. At that time, she began to receive treatment with antiepileptic drugs (AEDs). Her seizures, including myoclonic movements and GTCSs, were aggravated with oxcarbazepine, carbamazepine, and lamotrigine treatment, and significantly improved, particularly in GTCS, with valproic acid treatment. Frequency of GTCS was reduced to three or four times a year.
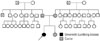
At the age of 20 years, she was admitted to our hospital due to aggravation of GTCS control (two times per month). She had been treated with levetiracetam (2000 mg/day), valproic acid (500 mg/day), clonazepam (0.75 mg/day), baclofen (20 mg/day), and lorazepam (1.5 mg/day). On neurologic examination, the almost continuous, tremulous movements of both hands were observed. Relatively large amplitude myoclonic movements occurred intermittently, particularly when moving suddenly, and were also provoked with arm stretching and hand extension. Also, she showed a mild degree of dysarthria, dysmetric limb ataxia, and truncal ataxia on tandem gait. The patient's 15-year-old sister had normal cognition and no neurologic symptoms. Her father had experienced several seizures after head trauma in his 30s. After taking AEDs for three years, seizures no longer developed. Also, this family had no ancestors from other countries. Other family history was unremarkable (Fig. 1).
Continuous video-electroencephalography (EEG) was performed. The EEG showed intermittent generalized sharp waves with posterior emphasis, although the myoclonic movements, including the almost continuous, small amplitude jerks, were not time-locked to EEG discharges. Background rhythm on EEG was normal with well-regulated 10-Hz posterior dominant rhythm. Photic stimuli could not provoke electrical and clinical seizure. Brain magnetic resonance imaging and magnetic resonance spectroscopy revealed no abnormalities. Somatosensory-evoked potential and visual-evoked potential tests showed normal latencies, shapes, and amplitudes.
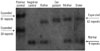
CSTB gene-associated dodecamer repeat expansion was analyzed by Southern blot analysis after obtaining informed consent. Genomic DNA was extracted from peripheral blood leukocytes using the Wizard Genomic DNA Purification kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. Southern blot analysis was performed at Athena Diagnostics Inc. (Marlborough, MA, USA). The patient showed only abnormally enlarged restriction fragments of 62 dodecamer repeats, confirming ULD, that were transmitted from her father and mother who carried the abnormally enlarged restriction fragments as heterozygotes with normal-sized fragments (Fig. 2). Her sister, also a heterozygote, carried the expanded fragment as a carrier.
We changed AEDs for seizure control. Upon discontinuing clonazepam, baclofen, and lorazepam, the patient was treated with valproic acid (1250 mg/day), levetiracetam (2000 mg/day), clobazam (10 mg/day), and zonisamide (300 mg/day). GTCS did not recur in the following 9 months. Further, her small amplitude jerks of both hands much decreased.
DISCUSSION
The present patient's clinical manifestations were compatible with PMEs related to several etiologies. ULD has rather milder symptoms than other PMEs. Our patient experienced gradually worsening, but mild neurological symptoms and preservation of normal EEG background activity. Therefore, these clinical features and the disease course of our patient were regarded as the typical clinical manifestations of ULD. If there is a severe progression in cognitive or visual symptoms, other forms of PME, such as myoclonic epilepsy with ragged red fibers, neuronal ceroid lipofuscinosis, Lafora's disease, and sialidosis, should be suspected.7 Recent study suggests that longer CSTB expansion mutations have a modulating effect on age at disease onset, myoclonus severity, and cortical neurophysiology in ULD.5 Abnormal CSTB gene-associated dodecamer repeat expansion was confirmed by Southern blot, leading to the diagnosis of ULD. Normal alleles of the CSTB gene contain two to three dodecamer repeats and full-penetrance alleles of more than 30 repeats.8 The size of fully expanded alleles and GC-rich sequences in the dodecamer repeat region make conventional PCR extremely difficult and require laborious Southern blot analysis. More convenient diagnostic testing (the dodecamer repeat analysis method, such as repeat PCR assay) needs to be developed in the future.
Although the prevalence of ULD is high in the Mediterranean and Finland, sporadic cases have been diagnosed worldwide, including Japan.59 However, the recognition of ULD is still lacking in areas of low prevalence due to varying availability of molecular diagnostic techniques and since it is difficult to diagnose ULD with clinical symptoms alone.10 To our knowledge, our patient is the first case to be confirmed genetically in Korea. In patients with intractable myoclonus or myoclonic epilepsy, especially with onset after the age of 6 years, analysis of the CSTB gene using Southern blot analysis will help to define the syndrome. Also, adequate choices of AEDs might be important in treatment of symptoms in ULD. Carbamazepine, oxcarbazepine, and lamotrigine may paradoxically worsen myoclonus as seen in our patient. An inverse pharmacodynamics effect is suggested as the main mechanism of aggravation of myoclonic seizures.11
 XML Download
XML Download