

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Pancreatic cancer, one of the most lethal human tumors, is characterized by aggressive malignancy and high resistance to traditional chemotherapeutic agents. The overall prognosis of patients with pancreatic cancer remains poor, with only 7% of patients living at 5 years after diagnosis.12 Most patients present with locally advanced or metastatic disease. Fewer than 20% of patients present with localized tumors indicated for potential curative surgical resection, and the majority of patients experience early local recurrence and metastasis.23 Gemcitabine remains the chemotherapeutic agent of choice for the treatment of pancreatic cancer. However, treatment with gemcitabine is associated with a response rate of less than 20%.45 Thus, a better understanding of the underlying molecular mechanisms of malignant progression is needed for targeted therapies inhibiting key cell signaling molecules associated with pancreatic tumor growth for improved patient outcomes.24
Targeted therapies have been developed to inhibit activated pathways that play a major role in cancer pathogenesis. One such therapeutic target is SRC, a non-receptor tyrosine kinase that belongs to the SRC family kinases (SFK). SRC induces cell proliferation, survival, cellular transformation, angiogenesis, invasion, and metastasis.678 SRC also plays a critical role in integrating divergent signal transduction pathways via interactions with multiple proteins. Activated downstream signaling pathways include heterotrimeric G proteins, mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT), signal transducer and activator of transcription 3 (STAT3), focal adhesion kinase (FAK), and c-MYC/cyclin D1 pathways.7910 The expression and activity of downstream signal proteins, as well as SRC, are correlated with cancer progression, malignancy, and poor prognosis in a variety of human cancers, including pancreatic cancer.101112 These findings, collectively, suggest that SRC and its signal proteins are attractive therapeutic targets.
Recently, AKT has emerged as an important protein and a potentially effective target for cancer therapy.1013 AKT is a serine/threonine protein kinase, and its activation controls cell growth, transformation, differentiation, motility, and survival.14 The PI3K/AKT pathway mediates cancer development and resistance to apoptotic effects of chemotherapy in different cancer types.14151617 AKT signaling pathways are considered attractive molecular targets in pancreatic cancer therapy due to their significant activation and involvement in pancreatic cancer cell growth.1819
It is essential to determine the efficacy of different combinations of signal inhibitors to overcome the challenges associated with pancreatic cancer treatment. In the present study, the anti-cancer effect of combined therapeutic targeting of signal molecules in pancreatic cancer cells was studied. Based on previous observations suggesting that SRC and AKT are interactive signaling targets, we evaluated the inhibitory effect of SRC combined with AKT on the proliferation, growth, migration, and anchorage-independent colony formation of pancreatic cancer cells in vitro and in vivo.
MATERIALS AND METHODS
Materials
Fetal bovine serum (FBS), trypsin, antimycotic antibiotics, and Dulbecco's Modified Eagle's Medium (DMEM) were purchased from GenDEPOT (Barker, TX, USA). 3-(4-chlorophenyl) 1-(1,1-dimethylethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (PP2) and 10-[4′-(N,N-Diethylamino)butyl]-2-chlorophenoxazine (10-DEBC) were obtained from Tocris Bioscience (Bristol, UK). DNase and RNeasy Mini Kits were purchased from Qiagen (Hilden, Germany). TRIzol and Lipofectamine RNAiMAX were ordered from Invitrogen (Carlsbad, CA, USA). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethylsulfoxide, and protease inhibitor cocktail were purchased from Amresco (Solon, OH, USA). Mammalian Protein Extract Reagent and Coomassie Plus Protein assay were supplied by Thermo Scientific (Rockford, IL, USA). Primary antibodies against mammalian target of rapamycin (mTOR), p-mTOR, extracellular signal-regulated kinase (ERK), and p-ERK were purchased from Cell Signaling (Boston, MA, USA), and the primary antibody against β-actin was obtained from Santa Cruz Biotechnology (Dallas, TX, USA).
Cell culture
The human pancreatic cancer cell lines MIA PaCa-2 and PANC-1 were obtained from the Korean Cell Line Bank (Seoul, Korea). The MIA PaCa-2 and PANC-1 cells were maintained in DMEM supplemented with 10% FBS and 1% antimycotic antibiotics. The cells were grown at 37℃ in a humidified atmosphere of 95% air and 5% CO2. The culture media were exchanged every 2 days.
Cell proliferation assay
Cell proliferation was determined by MTT assay.20 Pancreatic cancer cells were diluted with culture medium to a seeding density of 5×103 cells/well, plated on 96-well flat bottom plates, and incubated at 37℃ overnight. The cells were treated with chemical inhibitors. After incubation for 72 h, 10 µL of MTT solution (5 mg/mL) was added to each well, and the plates were incubated for another 4 h. After incubation, 100 µL of dimethylsulfoxide was added to each well to solubilize the dark blue MTT formazan product under gentle shaking. Luminescence was quantified using a microplate reader (VICTOR3, PerkinElmer, Waltham, MA, USA). Absorbance was measured at 560 nm, relative to the reference values at 670 nm, to represent the number of viable cells. Growth inhibition was calculated as the percentage of viable cells, compared with untreated cells (control group). Each assay was performed in triplicate.
Western blotting analysis
Pancreatic cancer cells were lysed in Mammalian Protein Extract Reagent containing a protease inhibitor cocktail. After sonication, the samples were centrifuged at 12000×g for 10 min at 4℃. Protein content in the supernatants was measured with Coomassie Plus Protein assay using bovine serum albumin as a standard. The proteins were mixed with the sample buffer, boiled for 10 min, and separated by 10–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The gel was transferred to a polyvinylidene difluoride membrane and blocked with Tris-buffered saline containing Tween 20 (TBST, 20 mM Tris-HCl, pH 7.6, 137 mM NaCl, 2.7 mM KCl, 0.05% Tween 20) and 3% bovine serum albumin for 1 h at room temperature. The membrane was incubated with primary antibodies against mTOR, p-mTOR, ERK, or p-ERK at 4℃ overnight (1:1000), washed with TBST three times, and incubated with anti-IgG secondary antibodies (1:2000) in TBST for 2 h. After the membrane was washed with TBST three times, the protein bands were visualized using EZ-Western Lumi Pico (Daeilab Service, Seoul, Korea). Protein loading was compared by probing the blots with anti-β-actin antibody. The membranes were stripped by Easter-Blot™ Western blot Stripping buffer (Biomax, Seoul, Korea) and reused.
siRNA inhibition assay
The target messenger RNA (mRNA) was knocked down using small interfering RNA (siRNA) as previously described.621 Briefly, the siRNAs (Bioneer, Daejeon, Korea) against human SRC, AKT1, AKT2, and AKT3 were used. Cells were grown at 70% to 80% confluence overnight, followed by transfection with predesigned siRNA (50 nM) using Lipofectamine RNAiMAX transfection reagent as instructed by the manufacturer. No-target siRNA (scrambled siRNA) was used as a negative control. After 48 h, the cells were assayed. The expression of SRC, AKT1, AKT2, and AKT3 was detected by quantitative RT-PCR and/or Western blot to demonstrate successful silencing of targets. The siRNA decreased mRNA expression of SRC, AKT1, AKT2, and AKT3 by more than 80% and their protein expression by more than 70% in pancreatic cancer cells.
Cell migration assay
Cell migration was measured by wound assay.6 The pancreatic cancer cells were grown on 60-mm tissue culture dishes until confluent. Separate wounds were generated by scratching the monolayer of cells using a sterile plastic micropipette tip and rinsed gently with phosphate-buffered saline (PBS) once to remove non-adherent cells. Culture media containing chemical inhibitors were added. Wound closure was monitored by phase-contrast microscopy (Olympus, Tokyo, Japan), and digital images were obtained after 48 h of post-wounding.
Soft agar colony formation assay
A soft agar colony formation assay was used to assess the anchorage-independent growth ability of cells. Briefly, pancreatic cancer cells were harvested and suspended in DMEM containing 0.3% agarose and 10% FBS, and then plated on top of semisolid 0.5% agarose medium in 60-mm dishes (7500/dish). Cells were incubated at 37℃ in 5% CO2 for 14 days, and treated with chemical inhibitors every 3 days. Colonies grown on soft agarose were fixed by methanol, stained by 1% crystal violet, and counted with a microscope.
In vivo treatment
The xenotransplant model of pancreatic cancer was developed as previously described.21 This study was approved and conducted in accordance with the regulations and guidelines of the Institutional Animal Care and Use Committee at CHA University (Seongnam, Korea). BALB/c nude mice were housed in a light- and temperature-controlled aseptic environment at the Laboratory Animal Research Center in CHA University. Animals were purchased from OrientBio (Seongnam, Korea). BALB/c nude mice were acclimatized to laboratory conditions (20–22℃, 12 h/12 h light/dark, 40% to 60% humidity, and access to food and water ad libitum) for two weeks prior to experimentation. The MIA PaCa-2 and PANC-1 cells were collected and re-suspended in PBS. Approximately, 2×106 cells (100 µL) were injected subcutaneously into both flanks of 6-wk-old male BALB/c nude mice, and the xenotransplants were allowed to grow. When induced tumors increased to 5 mm in diameter, the mice were randomly divided into four treatment groups of five to six mice each. PBS, 10-DEBC, PP2, or 10-DEBC with PP2 was injected i.p. at 1 mg/kg once a week for a total of four times. Tumor size was measured once a week by calipers based on the following formula: tumor volume=(length×width2)/2 for 1 month. Tumor weight was measured after tumor tissue excision. Mice were monitored daily for signs of toxicity. No serious adverse effects were observed, such as weight loss, ruffling of fur, reduced life span, behavioral changes, and changes in feeding habit, during the experimental period.
RESULTS
Anti-proliferative effects of AKT and SRC inhibition on pancreatic cancer cells
The effects of AKT and SRC inhibitors on pancreatic cancer cell proliferation were investigated. Two human pancreatic cancer cell lines, MIA PaCa-2 and PANC-1, were treated with the AKT inhibitor 10-DEBC or SRC inhibitor PP2, and cell proliferation was measured after 72 h. Two inhibitors showed concentration-dependent anti-proliferative activities in the micromolar range of 0.1 µM to 300 µM in the two cell lines (Fig. 1A and B). The concentration of 10-DEBC that elicited a half-maximal response (IC50) for the inhibition of MIA PaCa-2 cell proliferation was 5.1±1.2 µM and that of PP2 was 31.3±1.1 µM (n≥6). In PANC-1 cells, the IC50 of 10-DEBC was 11.9±1.1 µM, while that of PP2 was 45.1±1.2 µM (n≥6). The two cancer cell lines showed similar sensitivities to the inhibitors (10-DEBC>PP2), and 10-DEBC yielded higher maximal response (3%), compared with PP2 (11–20%). This result indicated that the inhibition of AKT and SRC significantly attenuated the proliferation of pancreatic cancer cells.
To investigate the effects of combined targeted therapy, the MIA PaCa-2 and PANC-1 cell lines were exposed to 10-DEBC and PP2 at low concentration (1–3 µM) (Fig. 1C and D). Even though a single application had little effect on proliferation, the combined application of 10-DEBC (3 µM) and PP2 (3 µM) significantly suppressed the proliferation by 90.8±2.1% (p=0.001, n=9) in MIA PaCa-2 and 82.4±5.1% (p=0.001, n=9) in PANC-1 cells. These findings demonstrate that simultaneous inhibition of AKT and SRC induces synergistic anti-proliferative effects on pancreatic cancer cells.
Anti-proliferative effects of AKT and SRC knockdown on pancreatic cancer cells
The synergistic suppressive effects of 10-DEBC and PP2 were also demonstrated by knockdown of mRNA using specific siRNAs against AKT1, AKT2, AKT3, or SRC. Concurrent knockdown of AKT2 with SRC in pancreatic cancer cells induced a significantly larger reduction in proliferation, compared with other combinations (n=6, p=0.006 for MIA PaCa-2; p<0.001 for PANC-1) (Fig. 2). The simultaneous inhibition of AKT2 and SRC induces synergistic anti-proliferative effects against pancreatic cancer cells.
Inhibition of AKT and SRC suppressed pancreatic cancer growth in vivo
In vivo anti-tumor activity of the combined application of 10-DEBC and PP2 was measured using a xenotransplant model (Fig. 3). Tumor volume analysis was conducted once a week for a total of four times following i.p. injection of 10-DEBC (1 mg/kg), PP2 (1 mg/kg), or 10-DEBC with PP2 (Fig. 3A). One month after the first injection, tumor volumes in the PBS, PP2, and 10-DEBC-treated mice increased by approximately 10–15 times, compared with the tumor volume on day 0, while that of 10-DEBC/PP2-treated mice showed only about a 3- to 4-fold increase. The group exposed to combined 10-DEBC and PP2 treatment showed a significant difference from the control group in tumor size from day 21 to day 28 (MIA PaCa-2 cell, p=0.004 for day 21, p=0.007 for day 28; PANC-1 cell, p=0.013 for day 21, p=0.042 for day 28, n=7). The tumor weights of the four groups were analyzed on day 28 (Fig. 3B). Tumor weight following co-application with 10-DEBC and PP2 was 47.2±7.0% of the control group in MIA PaCa-2 and 44.4±6.4% in PANC-1, which was significantly lower than that in the control group (p=0.004, p=0.004, respectively, n=7). As shown in Fig. 3C, tumors from 10-DEBC/PP2-injected mice were the smallest among those tested. The simultaneous inhibition of AKT and SRC resulted in a synergistic anti-growth effect on pancreatic tumors.
Inhibition of AKT and SRC attenuated the metastatic potential of pancreatic cancer cells
Wound assays were performed to evaluate the effect of cotreatment with inhibitors on the migration of pancreatic cancer cells (Fig. 4A and B). Scratch widths were recovered by 50.5±2.8% in MIA PaCa-2 and 76.0±2.8% in PANC-1 after 2 days (n=6). Application of 10-DEBC (0.3 µM) with PP2 (1 µM) effectively reduced the migration of MIA PaCa-2 cells to 27.1±1.7% (p=0.005) and PANC-1 cells to 32.7±6.0% (p=0.005). In addition, the colony formation was reduced by inhibitors in pancreatic cancer cells (Fig. 4C and D). Treatment with a combination of 10-DEBC (0.3 µM) and PP2 (1 µM) significantly inhibited colony formation of MIA PaCa-2 cells by 92.2±2.3% (p=0.030, n=4) and that of PANC-1 cells by 82.1±2.5% (p=0.013, n=4). The suppression of metastatic potential, cell migration and colony formation, supported the synergistic inhibition of proliferation induced by co-treatment with 10-DEBC and PP2.
Signal protein expression
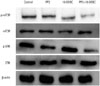
AKT and SRC are integral parts of several signal pathways, such as those mediating mitosis, invasion, and metastasis. To determine whether 10-DEBC and PP2 treatment modulate the expression of key signal proteins, mTOR and ERK, during the proliferation and metastasis of pancreatic cancer cells, MIA PaCa-2 cells were exposed to PP2 (3 µM) and 10-DEBC (3 µM) for 1 h, and the resultant protein expression was analyzed using Western blot (n=3) (Fig. 5). Exposure to PP2 and 10-DEBC markedly decreased p-mTOR and p-ERK expression in pancreatic cancer cells. A reduction in phosphorylation of mTOR and ERK may mediate the synergistic anti-cancer effect of 10-DEBC and PP2.
DISCUSSION
In preclinical studies, SRC inhibitors exerted significant anticancer effects on pancreatic cancer; however, clinical trials thereof in advanced pancreatic cancer have not shown a significant therapeutic benefit.22 Changes in therapeutic strategies for advanced pancreatic cancer should be considered. By additionally inhibiting SFK members and SRC signal partners, the therapeutic effectiveness of SRC inhibitors can be enhanced. Our previous study revealed the potency of SFK members as therapeutic targets using specific, targeted siRNA.6 In the current study, the SRC signal partner AKT was investigated as a concurrent target.
SRC plays a key role in cancer survival and progression via proliferation, migration, invasion, adhesion, and angiogenesis. The inhibition of SRC in pancreatic cancer cells by the specific chemical inhibitor PP2 reduced cell proliferation in a concentration-dependent manner, with an IC50 of 30–45 µM. In a previous study, additional SRC inhibitors, AZM475271 and A419259, showed similar anti-proliferative effects with IC50 values of 40–50 µM and 5–10 µM, respectively.6 PP2 also suppressed cell migration and agar colony formation by 10–40% (data not shown). In addition, SRC regulates the expression of interleukin (IL)-8 and vascular endothelial growth factor (VEGF) in angiogenesis, gemcitabine chemosensitivity, and the expression of insulin-like growth factor I receptor (IGF-IR) and matrix metalloproteinases in invasive cancer.2324252627 The antitumor effects of SRC inhibitors have been studied in preclinical models and in a clinical trial.222829
By regulating many aspects of oncogenesis, SRC acts as a master controller. SRC activates multiple targets and signaling pathways, such as Ras/MAPK, STAT3, PI3K/AKT, FAK, Cadherin, and annexin II.30 The key effects of SRC are mediated via activation of downstream signaling pathways, including the PI3K/AKT pathways. Potent or synergistic effects of SRC are required to reveal significant targets and downstream pathways. In the present study, inhibition of SRC in combination with AKT was investigated to evaluate significant co-targets in pancreatic cancer. Although the application of 10-DEBC or PP2 alone at low concentrations had little effect on cell proliferation, the application of 10-DEBC with PP2 significantly suppressed the proliferation by 80–90%. Further, the inhibition of AKT with SRC reduced tumor growth in xenotransplant models. Furthermore, this inhibitor combination exerted anti-migratory and anti-invasive effects in pancreatic cancer cells. Thus, the interaction between AKT and SRC may be important in cancer cell proliferation, growth, migration, and invasion. These results further support that AKT and SRC are potential concurrent targets in pancreatic cancer treatment. SRC regulates critical downstream signaling involving AKT. SRC activity regulates phosphorylation of AKT, p38, and ERK-1/2, which are involved in angiogenesis-associated IL-8 expression.23 The PI3K/AKT pathways are activated in a SRC-dependent fashion upon epidermal growth factor (EGF) receptor activation and EGF-mediated VEGF production.24 The suppression of SRC expression has been found to decrease AKT activity and increase gemcitabine chemosensitivity.25 SRC activity is essential for the up-regulation of IGF-IR by CEACAM6-induced AKT activity.31 AKT activated by active SRC leads to invasiveness of cancer cells via up-regulation of IGF-IR expression.26 Simultaneous inhibition of AKT and SRC may block signaling pathways involved in proliferation, angiogenesis, chemosensitivity, and invasiveness. In the present study, the inhibition of AKT along with SRC suppressed the phosphorylation of mTOR and ERK, which inhibited tumor growth and metastatic potential.
Several studies have investigated the concurrent inhibition of important therapeutic targets. STAT3 and EGFR were inhibited resulting in enhanced sensitivity,32 and SRC and EGFR were inhibited to overcome gemcitabine resistance in pancreatic cancer.33 SRC, STAT3, and MYC were targeted using siRNA resulting in maximal anti-cancer effect under a variety of cellular conditions.10 The concurrent targeting of signal proteins may result in higher efficiency, fewer side effects, and use of multimodal therapies, such as chemical and small molecule inhibitors, and siRNA.
Combined inhibition of AKT and SRC showed a synergistic effect at low concentration, although the inhibition of SRC or AKT resulted in anti-cancer effects. These results strongly suggest that the combined targeting of AKT and SRC may produce benefits exceeding those provided by SRC inhibition alone. These findings may facilitate significant improvement in the traditional treatment of pancreatic cancer and represent a breakthrough for the survival of patients with pancreatic cancer, with predominantly low survival rates. The present study may augment the anti-cancer effects of therapeutics targeting cell signaling pathways involved in tumor growth and metastasis.




 XML Download
XML Download