

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Guillain-Barré syndrome (GBS) is now known to cover a spectrum of postinfectious dysimmunogenic peripheral neuropathies.1 Recent advances in techniques for investigating the immunological mechanisms of GBS have revealed that various antiganglioside antibodies play pathogenic roles and influence its phenotypes.23 Various recent trials have classified and categorized this disease group based on immunological profiles (i.e., antiganglioside antibodies)4 or electrophysiological characteristics.5 There is emerging evidence that GBS is characterized by geographical variations in the incidence of each subtype.36
A syndrome of cranial mono- or polyneuropathies is one of the clinical variants of GBS in additional to either Miller-Fisher syndrome (MFS) or classical limb-dominant GBS.789 Identifying the antiganglioside antibodies in the sera of patients can act as a specific biomarker for categorizing each syndrome of GBS. This has led to several syndromes being defined based on the positivity to certain antibody types, such as anti-GM1, anti-GQ1, or even anti-GT1a syndrome.48 Neither immunoglobulin (Ig)M- nor IgG-type anti-GM2-ganglioside-antibody syndrome has been clinically characterized, which can be attributed to IgG- or IgM-type anti-GM2 antibodies being detected far less frequently in GBS than other types of antiganglioside antibodies. Additionally, a previous study found that GM2 could not be stained using an immunohistological technique.10 This situation has prompted many researchers to suspect that this syndrome plays a pathogenic role in human GBS.
Anti-GM2 antibodies—and especially the IgM type—have often been reported in the serum in acute and chronic dysimmune peripheral neuropathies as well as in motor neuron disease.11 The anti-GM2 reactivity in some of these patients was associated with a concomitant reactivity with GM1, while no reactivity with other gangliosides was detected in other patients, suggesting that GM2 is a target for autoantibodies in these patients.11
Rupp et al.12 recently identified GM2 in the canine sciatic nerve using both mass spectrometry and a thin-layer chromatography overlay technique. They additional used immunohistological methods to localize GM2 predominantly in the abaxonal Schwann cell membrane.
There is evidence from both in vivo and in vitro studies for the involvement of anti-GM2 antibodies in the development of acute immune-mediated peripheral neuropathies, such as GBS.101314 However, no previous study has analyzed the immunological-phenotypical correlation for anti-GM2 antibodies in GBS.
This study aimed to identify the incidence of GBS as defined clinically and electrophysiologically based on IgM- or IgG-type anti-GM2-antibody positivity. In addition, we attempted to characterize the clinical phenotypes and define the syndrome based on its unique characteristics relative to other antibody syndromes.
METHODS
Patients and clinical data
The Dong-A University Neuroimmunology Team (DAUNIT) has applied an antiganglioside antibodies assay to the sera of cases with a presumptive diagnosis of acute immune-mediated cranial or peripheral neuropathy. The Korean Inflammatory Neuropathy Consortium (KINC), which was established in 2012 as a nationwide society for inflammatory neuropathies, has systematically collected clinical and laboratory information on GBS from university-based hospitals in Korea. The collaboration between the DAUNIT and KINC means that the clinical and specific laboratory data of each case are collected from the on-site investigators at the respective institutions in the KINC.7 The records of each case were anonymized and de-identified prior to being analyzed.
This study reviewed 2109 consecutive cases after combining the DAUNIT and KINC databases obtained from 2007 to February 2017. The DAUNIT collected sera related to immune-mediated neuropathies and other acute cranial neuropathies from more than 40 general and university-based hospitals in Korea over a period of more than 10 years. Detailed information on the involved patients was often obtained and then corrected by the referring neurologists applying additional questionnaires. GBS was defined based on conventional clinical criteria15 with supporting results of anti-ganglioside-antibody positivity.16 Additionally, compatible specific variants of GBS such as MFS, the pharyngeal-cervical-brachial variant, and Bickerstaff brainstem encephalitis were defined based on a recently proposed classification system.4 The enrolled cases were analyzed and compared based on both clinical and objective findings such as laboratory and electrophysiological features.
Anti-ganglioside-antibody tests
All serum samples were obtained from patients within 2 weeks of symptom onset and collected by the DAUNIT for anti-ganglioside-antibody tests. An enzyme-linked immunosorbent assay was used to detect IgG- and IgM-type antibodies against the gangliosides GM1, GM2, GD1a, GD1b, GD3, GT1a, GT1b, and GQ1b according to previously described methods with some modifications.1617 The presence of different types of antiganglioside antibodies was analyzed by researchers who were blinded to the clinical information of the patients.
RESULTS
The data set from antiganglioside antibody assays performed from 2007 to February 2017 by the DAUNIT was reviewed. As mentioned above, the systematic and prospective enrollment of both clinical and laboratory data started in 2012, and hence it is necessary to address the presence of heterogeneity and possible contamination by diseases other than GBS in our cohort, such as idiopathic oculomotor palsy, facial diplegia, or bulbar palsy.
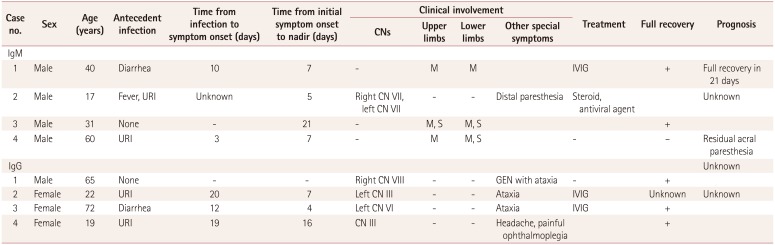
We reviewed the laboratory results of the 2019 included patients, which revealed 458 cases that were positive for any of the antiganglioside antibodies studied (22.7%). Ten of these patients were positive for either isolated IgM- or IgG-type anti-GM2 antibody (2.2%), while they were negative for antibodies other than anti-GM2 antibodies. Two of these 10 patients were excluded after repetitive history-taking: both patients showed clinical progress over a 1-month period and were finally diagnosed as chronic idiopathic peripheral neuropathy. The eight enrolled patients shared the following common features of GBS: monophasic course and recovery, taking <4 weeks to clinical nadir, and absence of etiologies of other peripheral neuropathies.
Finally, four patients were identified as classical limb-dominant peripheral polyneuropathy or cranial neuropathy with isolated IgM-type anti-GM2-antibody positivity. Another four patients had acute neuropathies, with only IgG-type anti-GM2 antibody in their sera. Therefore, finally 8 patients with GBS were identified as an anti-GM2-ganglioside-antibody syndrome with isolated positivity of either the IgM or IgG type from among the 2019 analyzed cases (0.4%).
The demographic and clinical features of the enrolled patients are summarized in Table 1. The mean age at onset was 41 years (range, 17–65 years), and the subjects comprised five men and three women. Both the IgM- and IgG-type anti-GM2-antibody groups had positive histories for various preceding infections: upper respiratory infection (two out of four, 50%), diarrhea (one out of four, 25%), and no infection history (one out of four, 25%). The latency of the first symptoms after infection, pattern of involvement, and disease course varied between the patients. One patient in the IgM group and two in the IgG group were treated with a 5-day course of intravenous Ig.
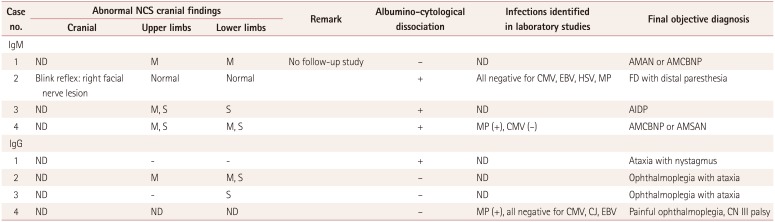
Table 2 lists the electrophysiological and laboratory findings of the eight patients with GBS and its variants with either IgM- or IgG-type anti-GM2-antibody positivity. The enrolled patients were unfortunately not fully evaluated for possible infections. Two of the four patients with IgM and one of the four patients with IgG underwent serum tests for Mycoplasma pneumoniae infection. Only one patient in each of the IgM and IgG groups was identified as having recent Mycoplasma pneumoniae infections. Only three patients underwent serological testing for cytomegalovirus (CMV) infection, and the findings for all of them were negative.
Albuminocytological dissociation was present in three of the four patients (75%) in the IgM group but only one of the four patients (25%) in the IgG group. Various abnormalities were identified in conventional nerve conduction studies in both groups.
The clearest overall difference between the two groups was in the pattern of regional involvement of peripheral nerves. The IgM-positive group comprised heterogeneous syndromes: one of acute motor axonal neuropathy or acute motor conduction-block neuropathy (AMCBNP), one of AMCBNP or acute motor sensory axonal neuropathy, one of acute inflammatory demyelinating polyneuropathy (AIDP), and one of isolated facial diplegia. Thus, one patient manifested with facial diplegia while the other three patients manifested with limb peripheral neuropathy.
In contrast, all of the enrolled cases in the IgG-positive group manifested with dizziness, and three of them (75%) experienced ataxia. It appeared that more than one cranial nerve was involved in all of the patients with IgG-type anti-GM2-positive GBS: two patients had oculomotor nerve involvement (50%), one patient had abducens nerve involvement (25%), and one patient had probable involvement of the vestibular nerve (25%). All of the patients in both groups (with the exception of a single patient with residual acral paresthesia) showed full recovery after their monophasic illness.
DISCUSSION
This study found that patients with isolated anti-GM2-positive GBS were extremely rare in a cohort with acute immune-mediated or idiopathic peripheral neuropathies (8 of 2019 patients, 0.4%). Furthermore, the clinical manifestations were not consistent and difficult to categorize uniformly based on existing criteria.4 Two patients who were initially enrolled in this study were finally excluded because their clinical course was more compatible with chronic immune-mediated peripheral neuropathy than GBS. Our study therefore suggests that the anti-GM2 antibody is not specific to GBS. This issue needs to be elucidated by assessing the clinical-immunological correlation in larger numbers of patients with this rare syndrome.
Four of our patients with IgM anti-GM2 antibodies and four with IgG anti-GM2 antibodies showed a distinctive feature that differentiated the two groups, although the number of patients was too small to definitively define the clinical characteristics of a particular GBS variant. The IgM-type anti-GM2-positive GBS patients showed heterogeneous manifestations, whereas all four GBS patients in the IgG-type anti-GM2-positive group showed clinical features involving the cranial nerve. Three of the four IgM-type anti-GM2-antibody-positive GBS patients were categorized as motor-dominant or sensorimotor polyneuropathy, while all four patients with the IgG type had cranial involvement, with three showing either painful or painless ophthalmoplegia. One patient manifested with the so-called nystagmus-and-ataxia pattern.18 These findings are especially interesting in that most of the patients would have been misdiagnosed as another kind of cranial neuropathy or acute vestibulopathy.
The aforementioned weak clinical-immunological correlation means that whether or not anti-GM2 antibody also has a pathogenic role in the development of GBS, such as the “molecular mimicry” mechanism, cannot be concluded from the results of this study. These results can be explained by speculating that minor differences in the specificities of antibodies to the same antigenic target produce marked differences in clinical presentations and the regional involvement.
The exact amount and relative distribution of GM2 in the human peripheral nervous system is unknown. One study identified that GM2 can be found in all of the cranial nerves and both dorsal and ventral roots of the spinal nerves, albeit at a much lower concentration than other major gangliosides such as GD1a, GD1b, LM1, and GT1b.19
Various patterns of peripheral neuropathies associated with anti-GM2 antibody have been described previously, such as sixth-cranial-nerve palsy,20 facial diplegia,21 and chronic sensory ataxic demyelinating polyneuropathy.22 However, these syndromes usually show positivity for other types of antiganglioside antibodies in addition to the anti-GM2 antibody.
Apart from GBS, anti-GM2 antibodies in conjunction with anti-GalNAc-GD1a antibodies have also been linked to chronic demyelinating polyneuropathy with severe sensory ataxia in adults.22 A particularly interesting observation is that other ganglioside antibodies implicated in neuropathies tend to be the IgG type; however, all reported associations of anti-GM2 antibodies with GBS variants—including the cases mentioned here—are the IgM type.
Moreover, anti-GM2 antibodies have previously been shown to have specific relationships with CMV infection.2324 One study suggested that CMV patients with anti-GM2-antibody-positive GBS show severe sensory defects and cranial nerve involvement.23 Another study identified that CMV infection was the antecedent infection in 13% of cases and was clinically associated with severe sensorimotor impairment in GBS.25 In other words, the clinical phenotype of anti-GM2-antibody-positive GBS after CMV infection appears to be rather heterogeneous.
Ang et al.26 induced anti-GM2-antibody-recognizing epitopes in fibroblasts infected with CMV, revealing that viruses can induce the molecular mimicry of gangliosides and lending further credence to the molecular mimicry theory. They detected only IgM-type anti-GM2 antibody, but other studies have also detected IgG-type anti-GM2 antibody.1423
Cavanna et al.11 showed that anti-GM2 antibody can induce complement-dependent cytolysis. We therefore applied a cytotoxicity assay to neuroblastoma cells in sera from seven patients with demyelinating dysimmune neuropathies and high titers of IgM-type anti-GM2 antibody. However, anti-GM2 antibody can be detected from infectious agents other than CMV. One study found that 1 of 30 patients with GBS-/Fisher-syndrome-related mycoplasma infection showed IgM-type anti-GM2-antibody positivity.27 In addition, anti-GM2 antibodies and positive herpes simplex virus serology were reported in an adult patient with multiple cranial neuropathies, including external ophthalmoplegia.28
Another issue to consider is that GM2 cannot be detected in human peripheral nerves by the standard immunohistochemical techniques using IgM-type anti-GM2 antiserum.10 This raises some doubts about the pathophysiological significance of IgM-type anti-GM2 antibody in AIDP associated with CMV infection. It has even been reported that neurologically healthy humans occasionally also exhibit low anti-GM2 antibody titers.13 Nevertheless, some animal studies have pathologically identified that anti-GM2 antibody and its complement drive segmental demyelination in acute canine polyradiculoneuritis.12
The present study adds significantly to the scarce information in the literature about the electrophysiological characteristics of GBS, which includes one study suggesting that GBS after CMV infection is the demyelinating type.29
Some limitations of the present study should be considered. One issue is associated with sharing of epitopes by GM2 and other types of gangliosides, which was not tested in our cases. Lopate et al.22 reported that the antibody binding site—the terminal GalNAc(14)Gal(2-3)NeuAc trisaccharide moiety—might be shared by GM2 and GalNAc-GD1a gangliosides, as well as GalNAc-GD1b (which was not tested). We therefore cannot exclude the possibility of cross-reaction between various types of each antiganglioside antibody, though we did select cases with isolated anti-GM2-antibody positivity. Moreover, even though the DAUNIT database includes chronic immune-mediated peripheral neuropathies, we did not evaluate chronic cases of anti-GM2 antibody in this study. Finally, only one of the patients enrolled in this study was evaluated for recent CMV infections, and serological tests for mycoplasma infections were seldom performed in our cohort. All of these limitations should be considered when interpreting the present results.
We have identified that IgM- and IgG-type anti-GM2 antibodies can be found in various subtypes of GBS and its variants. IgG-positive patients can be characterized by cranial-dominant GBS variants presenting mainly with oculomotor and vestibular dysfunctions, while IgM-positive patients can manifest with heterogeneous syndromes.
 XML Download
XML Download