

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cerebellar ataxias (CAs) are a clinically, pathologically, and etiologically heterogeneous group of disorders.12 CAs can be commonly categorized into acquired, genetic, and sporadic degenerative ataxias.3 For patients with acute or subacute disease onset, initial investigations usually readily identify a symptomatic cause; thus, diagnosis of acquired ataxic disorders is relatively easy.14 However, patients with chronic progressive CA may present with widely variable and overlapping symptoms and signs, either as a pure cerebellar syndrome, or associated with significant cognitive, pyramidal, extrapyramidal, sensory, and autonomic dysfunction. Genetic CA is classified according to its inheritance patterns as follows: autosomal-dominant (AD), autosomal-recessive (AR), X-linked, or mitochondrial form.5 The sporadic degenerative CAs include the cerebellar variant of multiple-system atrophy (MSA-C) that is a pathologically and clinically well-defined entity, and sporadic adult-onset ataxia of unknown etiology (SAOA), which is distinct from MSA-C. Likewise, classifying CAs is complicated, and their investigation often poses considerable diagnostic challenges despite rapid advances in molecular genetics and imaging techniques, and many familial cases as well as most sporadic patients remain undiagnosed.3
The prevalence and frequency of CA subtypes vary between countries.6789101112 Although it is necessary to estimate the frequencies of subtypes when planning a diagnostic strategy in a specific population, studies of the epidemiology in all categories of CAs are surprisingly rare in Korea. Our primary aim in this study was to determine the frequency of each diagnostic group (familial and sporadic) in a movement-disorders outpatient clinic at a tertiary referral center.
METHODS
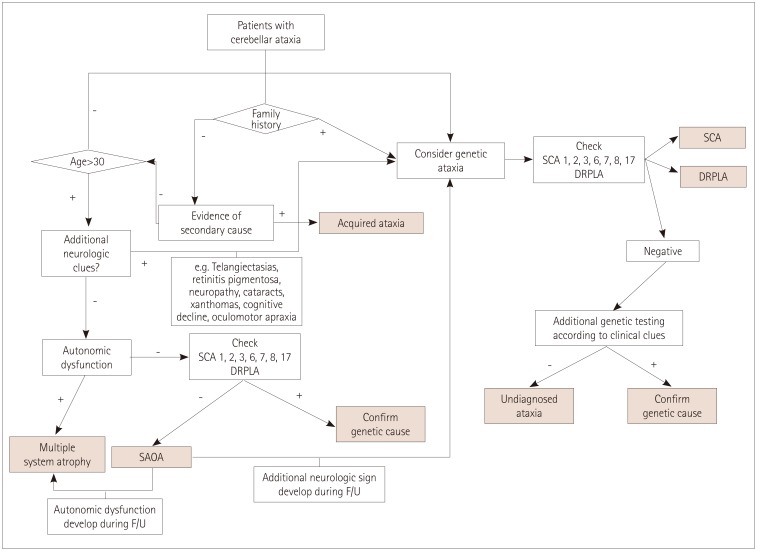
Patient selection and diagnostic workup (see flow chart)
The Movement Disorders Clinic in the Neuroscience Center, Samsung Medical Center (Seoul, Korea) is one of the largest centers of its kind in Korea and has acted as a referral center for patients with presumed ataxias since 1994. Among the patients who visited the Movement Disorders Clinic between November 1994 and February 2016, 2006 patients were clinically screened and examined as having suspected CA. Most patients were followed up on a 3-monthly basis, and those without a clear diagnosis were reassessed with repeated imaging and additional tests for confirming rare causes of ataxias. Patients who were followed up for at least 3 years were included. More than half of the patients visited our clinic once or twice to obtain a second opinion and were lost to follow-up. Patients with an incomplete assessment due to incomplete investigations or a very short period of follow-up were also excluded. The following information was collected: sex, age at symptom onset, time from disease onset to visiting our clinic, disease duration from symptom onset, and family history. The family history was collected based on available information about the presence of similar disorders or a history of unexplained gait disturbances in first- and second-degree relatives. We performed the following tests to detect acquired ataxias in patients whose progression was acute or subacute: thyroid function test, levels of vitamins B12 and E, serology for syphilis and HIV, autoimmune antibodies, tumor markers, paraneoplastic antibody (available since 2005 in our clinic), and brain magnetic resonance imaging. Patients who were already diagnosed with cerebral ischemia, hemorrhage, tumors, or demyelinating disease were excluded. Patients were evaluated with Korean ataxia genetic screening for disorders including spinocerebellar ataxias (SCA 1, 2, 3, 6, and 7) and dentatorubral-pallidoluysian atrophy (DRPLA) when they had 1) a family history, 2) a young onset age (<30 years), or 3) additional neurological clues (Fig. 1). Beginning in 2012, this evaluation was expanded to include SCA 8 and 17 due to their frequent detection in the Korean population. More specific genetic tests were performed in selected patients who were suspected as having a specific subtype of genetic ataxic disorder (e.g., patients with polyneuropathy or myopathy, telangiectasia, xanthoma, cognitive decline, oculomotor apraxia, or abnormal findings on a fundus examination). This study was approved by the Institutional Review Board of Samsung Medical Center (IRB No. 2013-12-055).
Categories
We divided CA patients into familial and non-familial ataxia, and analyzed the frequency of each etiology. Patients were finally categorized into having genetic, sporadic, secondary, or suspected genetic, but undetermined (i.e., undetermined) ataxia. The genetic group included patients whose genetic mutations were confirmed by genetic sequencing. In the case of the sporadic group, we diagnosed MSA-C according to the second consensus criteria of MSA.13 If a patient had a stationary disease course without a secondary cause, familial history and dysautonomic symptoms, they were grouped as SAOA after at least 3 years follow up.3 Patients who had a family history of similar symptoms, young age at onset (age of onset <30) or adult-onset sporadic ataxia where a genetic cause was clinically suspected, but genetic screening or sequencing results were negative, were labeled as undetermined ataxia. The secondary cause group included patients who were determined to have acquired causes through intensive investigations.
Additional prospective screening for Friedreich's ataxia
Patients with undiagnosed ataxia who were still visiting our clinic were prospectively evaluated with Friedreich's ataxia (FRDA) genetic screening. Among 91 patients with undetermined ataxia, 66 patients who visited the Movement Disorders Clinic during the period from March 2015 to March 2017 and agreed to participate in additional genetic analysis were enrolled. Screening test of homozygous GAA trinucleotide expansion was performed using long PCR technique.
Statistical analysis
Statistical analyses for subject demographic characteristics were performed with SPSS Statistics (version 18.0, SPSS Inc., Chicago, IL, USA). The chi-square test and one-way analysis of variance were used to compare demographic features among the genetic, sporadic, and secondary groups.
RESULTS
In total, 820 of the original 2006 patients were finally included in this study. Of these, 136 (16.6%) patients had a family history of ataxia and the rest of the 684 cases did not. The number of patients with a history of early onset ataxias (under the age of 30) was 113 (13.8%).
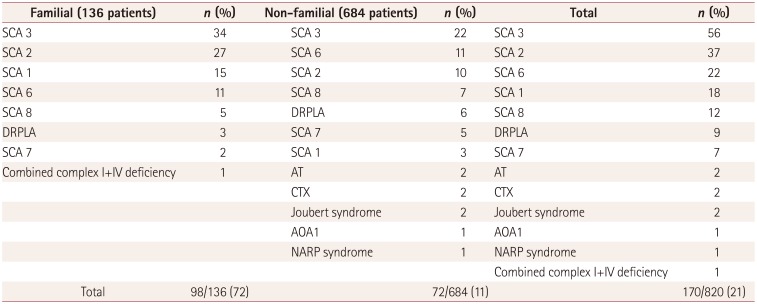
Table 1 reveals the frequencies of causative genes in familial and non-familial patients. Among all patients with familial ataxias, a genetic diagnosis was achieved in 98/136 (72%). 72/684 (11%) of non-familial patients had a confirmed genetic diagnosis even though they denied any family history. Finally, 170/820 (21%) patients were identified as having genetic disease.
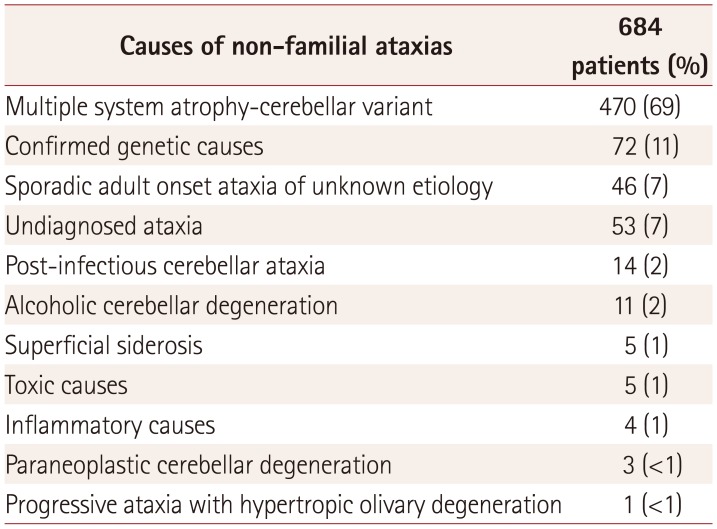
The frequency and causes of nonfamilial ataxias are described in Table 2. The most common cause of nonfamilial ataxia was MSA accounting for 470/684 (69%) cases, followed by confirmed genetic causes (72/684, 11%) and SAOA (46/684, 7%). A total of 53/684 (7%) patients had undetermined ataxia.
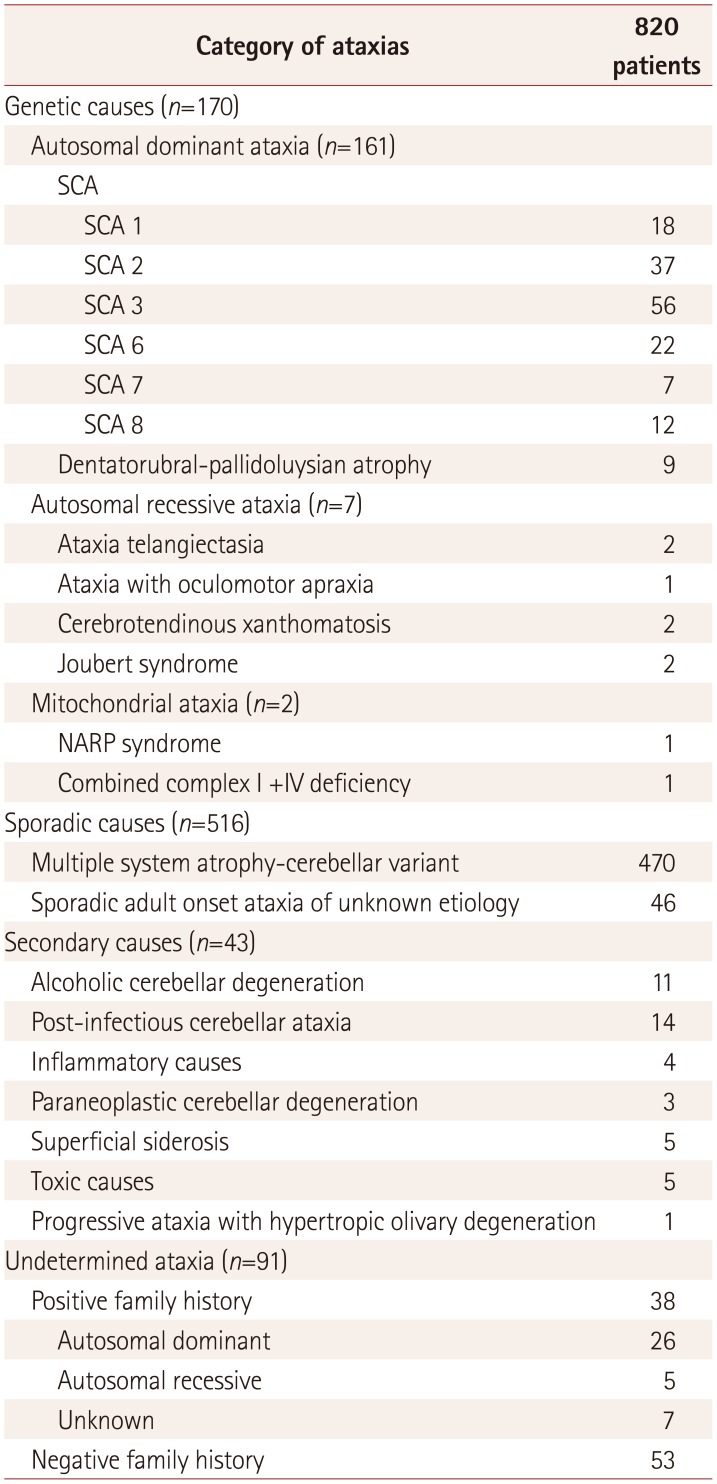
Table 3 summarizes the overall etiologies of the 820 cases of progressive ataxia in this study: 170/820 (21%) patients were genetic, 516/820 (63%) were sporadic, 43/820 (5%) were secondary and 91/820 (11%) were undetermined. In the genetic group, the most common etiology was SCA (152/170, 89.4%). The most common subtype of SCA was SCA 3, followed by SCA 2 and SCA 6. Nine cases of DRPLA (9/170, 5.3%) were identified. There were seven cases with AR ataxia, including ataxia telangiectasia, ataxia with oculomotor apraxia, cerebrotendinous xanthomatosis, and Joubert syndrome. Three cases were suspected as having mitochondrial ataxia, and neurogenic muscle weakness, ataxia, and retinitis pigmentosa syndrome, and combined complex I+ IV deficiency were confirmed in these cases. There were seven patients with a clinical history of episodic ataxia and a negative family history, but CACNA1A gene mutations were not detected in these patients.
In the sporadic group, 470 cases of MSA-C (470/516, 91%) and 46 cases of SAOA (46/516, 9%) were identified. The most common secondary cause was postinfectious CA, followed by alcoholic cerebellar degeneration.
Thirty-eight of the 136 familial cases and 53 of the 684 nonfamilial cases (total 91/820, 11%) remained as undetermined ataxia. A screening test for FRDA was performed in 66 of the patients with undetermined ataxia, and abnormal expansion of GAA repeats in the FXN gene was not found. The demographic characteristics of these patients are shown in Supplementary Table 1 (in the online-only Data Supplement).
Comparison of demographic characteristics in the genetic and sporadic groups
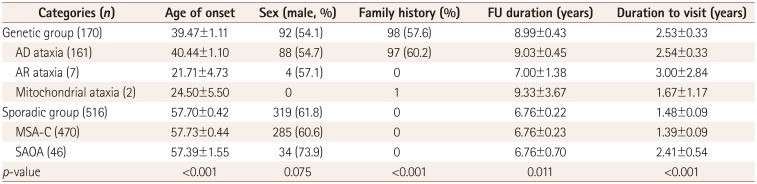
Table 4 presents the demographic characteristics, family history, follow-up duration (from the initial to the last visit), and time to diagnosis (from symptom onset to diagnosis) in the genetic and sporadic groups. The age at onset was significantly older in the sporadic group (57.70±0.42 years, mean±SE) than in the genetic group (39.47±1.11 years) (p< 0.001). Among patients in the genetic group, the age at onset was oldest in patients with AD ataxia (40.44±1.10 years), followed by mitochondrial (24.50±5.50 years) and AR (21.71±4.73 years) ataxia. The age at onset did not differ between patients with MSA-C (57.73±0.44 years) and SAOA (57.39±1.55 years). There was no difference in the sex ratio between the genetic and sporadic groups. In total, 98 (57.6%) patients in the genetic group had a family history. Only 97 (60.2%) of the patients with AD ataxic disorders had a family history. All of the patients with AR ataxic disorders denied having any family history. The mean follow-up duration to the last visit was 8.99±0.43 years in the genetic group and 6.76±0.22 in the sporadic group. The duration from disease onset to the initial hospital visit was longer in the genetic group (2.53±0.33 years) than in the sporadic group (1.48±0.09 years).
Forty-six patients were considered to have SAOA after a relatively long follow-up (mean follow-up duration 6.76±0.70 years). None of the patients developed autonomic dysfunction, and the ataxic symptoms also did not progress rapidly. For example, in a 76-year-old man diagnosed with CA 14 years ago, dysarthria, dysphagia and gait imbalance have progressed very slowly. However, he did not exhibit urinary dysfunction or orthostatic hypotension until now and his activities of daily living are well preserved.
DISCUSSION
Despite the fact that the frequency of the specific subtypes of CA differs considerably among different populations, there is a lack of objective studies exploring the distribution of CA types in the Korean population. The present study, which is the first of its kind in Korea, had revealed the various etiologies and frequencies of CA types in patients from a tertiary referral center. Although the data presented here are not representative of the Korean population, they are still noteworthy because this type of study will be helpful in planning diagnostic strategies and estimating the frequency of each subtype in the Korean population considering the large number of CA patients.
The MSA-C patients constituted the largest proportion of ataxic diseases. A German group1415 found only 32 patients with MSA-C among 466 patients in an ataxic population, whereas MSA-C was much more common in Asian areas such as Japan and Singapore, occurring in 67% of patients.16 Although no study has explored the exact prevalence of MSA-C, it may be the most common etiology of chronic progressive ataxia in Korea. SAOA is known to involve isolated degeneration of the cerebellar cortex with secondary atrophy of the inferior olives, a pattern of degeneration that was previously described as cerebello-olivary degeneration.17 Some of the patients in our study whose initial diagnoses were SAOA developed autonomic dysfunction after 2–3 years of follow-up, and were redesignated as MSA-C. However, 46 patients (9% of the sporadic group) were still categorized as having pure cerebellar syndrome and showed a benign disease course after a relatively long follow-up period (mean follow-up duration 6.76±0.22 years). Although the etiology and pathogenesis of SAOA are still unknown and it is not clear if this is a distinct disease entity or a collection of different but phenotypically similar diseases, SAOA with a benign disease course is distinct from MSA-C.318 Sporadic ataxia should therefore be diagnosed by careful ruling out and follow-up.
SCA 3 was the most common subtype in the genetic group, followed by SCA 2 and SCA 6. SCA 3 was found to be the most frequent subtype in Western Europe, North America, Brazil, and China, and has been estimated to be the cause of ataxia in 21% of patients worldwide.192021 According to previous Korean reports, SCA 2 or SCA 3 were frequent subtypes in the Korean population.522232425 There were considerable differences in the subtype frequencies of SCA among regions even in Korea. Our center is located in a central region, and our results are consistent with previous reports.
We identified nine cases of DRPLA (comprising 5% of the genetic group). Although DRPLA is generally very rare except in Japan, it is not uncommon in Koreans. We detected only seven (comprising 4% of the genetic group) cases with a recessive ataxia and no cases with an X-linked ataxia. Although the AR ataxias such as ataxia telangiectasia or FRDA are prevalent in most countries, they are extremely rare in the Korean population.26 In addition, genetic screening did not detect FRDA in the patients with undetermined ataxia. Therefore, routine screening of the FTX gene in the Korean population might not be needed unless patients show a clinical phenotype of FRDA. According to a recent review of fragile X syndrome epidemiology, the estimated frequency of individuals with the FMR1 allele premutation in the total population is approximately 1:850 for males and 1:300 for females. However, there has been only one report of fragile-X-associated tremor/ataxia syndrome in Korea, and X-linked ataxia is also rare in the Korean population.5
Thirty-eight of the 136 familial cases and 53 of the 684 sporadic cases (total 91/820, 11.1%) remained as undetermined ataxia. These patients had a family history or showed unique features such as polyneuropathy, retinitis pigmentosa, hearing loss, relatively young adult onset (<30 years), or restlesslegs syndrome, and did not meet the criteria for MSA and SAOA. While we routinely screened for the more common subtypes including SCA 1, 2, 3, 6, 7, 8, and 17 and DRPLA, tests for the less-common SCA mutations such as SCA 5, 10, 11–14, 17, 27, and 28 are not commercially available in Korea. It is possible that known gene mutations that were not tested for or are presently unknown gene mutations may underlie some of the unexplained cases.
An uncertain family history is a matter of great concern in the diagnostic process for genetic ataxia. Seventy-two (42% of the genetic group) patients in the genetic group denied having any relevant family history (Table 1). Recent advances in molecular genetics have indicated that up to 19% of sporadic cases may have a genetic basis.1127 This relatively large proportion of cases suggests that patient history was not taken accurately enough, however, the death of gene carriers before disease onset, offspring that have not reached the age of onset, reduced penetrance, marked anticipation, or de novo mutations could explain the lack of family history.2829 Owing to the wide spectrum of genetic causes, clinical heterogeneity, and uncertain family history, the correct diagnosis of ataxias remains a clinical challenge.
When we compared the demographic characteristics and time points between the genetic and sporadic groups, onset was younger in the genetic group compared to the sporadic patients. The time from disease onset to the first evaluation for diagnosis was longer in genetic patients than sporadic patients. This might indicate that the progression of genetic ataxia is slower than that of sporadic ataxia. Another possible explanation might be that patients with MSA-C felt more psychological discomfort when their symptoms first developed, due to the absence of a family history and the strikingly fast symptom progression.30
Our study had some limitations. First, we did not have pathological data for our patients, and so we cannot exclude that our patients actually had relevant pathology according to a clinical diagnosis. Second, we did not cover all of the AR ataxic disorders. However, cases with compatible AR ataxia phenotypes were not found and we performed specific gene sequencing in each case with a specific phenotype. But there is a possibility that we missed some genetic causes. Third, considering that our clinic is mainly focused on treating adult patients and that the onset of AR ataxia usually occurs during childhood, it is possible that many patients with AR ataxia were not included in this study. Actually, it is very likely given that the age at onset in patients with AR ataxia was 21.7 years in this study. Furthermore, secondary causes including stroke, multiple sclerosis, tumor, and inflammation would have been underestimated because our neuroscience center treats them separately. Fourth, we conducted FRDA gene screening in 66 of 91 patients with undetermined ataxia using the long PCR technique instead of full sequencing of the FRDA gene. So it is possible that we might lose patientswith FRDA. However, FRDA is extremely rare, and to the best of our knowledge has never detected in the Korean population. In addition, phenotypes of patients in whom FRDA screening test was performed, did not correspond with FRDA. Finally, because this was a single-center-based retrospective study, the reported data might not be representative of the entire Korean population.
In conclusion, this is the first study to analyze in detail the frequencies of CAs in a Korean population. The results will be helpful for clinicians who are planning diagnostic strategies and counseling for patients with CA. We have also provided further guidance and evidence that the SCA genes in patients with chronic progressive CA need to be screened regardless of their family history, even if the family history is less conclusive. Genetic technologies such as new-generation sequencing are currently being rapidly developed, and these advances will facilitate rapid progress in this field. In the future it will be important to explore the genetic background of undiagnosed patients in research projects investigating the less-common dominant and recessive ataxia genes.
 XML Download
XML Download