

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Chronic myeloid leukemia (CML) is a myeloproliferative disease characterized by a clonal disorder of hematopoietic stem cells. CML appears in 15–20% of all cases of leukemia, but it is very rare in children and adolescents, where it accounts for less than 2% of cases [12].
CML is genetically characterized by a reciprocal translocation that results in an exchange of genetic material between the long arm of chromosome 22 (22q−) and the long arm of chromosome 9 (9q+). This exchange is referred to as translocation t(9;22)(q34;q11.2) and creates a derivative chromosome 22 that is known as the Philadelphia chromosome (Ph). At the molecular level, this reciprocal exchange involves the Abelson oncogene (ABL1) on chromosome 9 and the breakpoint cluster region (BCR) gene on chromosome 22, which form a BCR-ABL1 fusion gene [3]. BCR-ABL1 exhibits a deregulated, constitutively active tyrosine kinase activity and is found exclusively in the cytoplasm of the cell complexed with a number of cytoskeletal proteins. Several functional domains have been identified in the BCR-ABL1 protein that may contribute to cellular transformation.
The standard treatment options for patients in the chronic phase of CML were previously hydroxyurea, interferon-α, or allogenic stem cell transplantation [4]. The treatment of CML was revolutionized in 2001 by the introduction of imatinib mesylate, which is a BCR-ABL1 tyrosine kinase inhibitor (TKI). The clinical use of specific BCR-ABL1 inhibitors has resulted in significant improvements regarding the prognosis, response rate, overall survival, and clinical outcomes of CML patients as compared to previous therapeutic regimens. The second-generation BCR-ABL1 TKIs, namely nilotinib and dasatinib, have shown significant activity in clinical trials in patients who are intolerant or resistant to imatinib therapy.
The Ph chromosome is detected in 90% of patients in the chronic phase of CML [5]. In the clinical diagnosis of CML, the ‘state of the art’ methods comprise classical banding techniques, fluorescence in situ hybridization (FISH), and real-time quantitative polymerase chain reaction (RQ-PCR) [6]. Besides the classical Ph translocation, it is possible to identify other patterns of chromosomal abnormalities in CML using the FISH method. Previously reported atypical patterns include variant translocations, the deletion of sequences proximal to the chromosome 9 breakpoint, the deletion of sequences distal to chromosome 22, and the deletion of the ABL1-BCR fusion gene [7].
The aim of this study was to analyze the frequencies of variant translocation and deletion on derivative chromosome 9 (del/der(9)) in newly diagnosed CML patients in a Croatian population. The complete cytogenetic response (CCyR) and the major molecular response (MMR) were also estimated at 12 months after the onset of TKI therapy.
Go to : 
MATERIALS AND METHODS
Materials
A retrospective study was conducted from January 2013 to October 2016 (total duration 3 yr and 9 mo) at University Hospital Centre Zagreb. Samples from 122 newly diagnosed Ph-positive CML patients were included. These patients comprised 53 (43%) women and 69 (57%) men (P=0.745). For cytogenetic analysis, each patient's bone marrow (BM) sample was taken in a test tube with lithium heparin anticoagulant. For monitoring MMR by RQ-PCR, a sample of peripheral blood (PB) with tripotassium ethylenediaminetetraacetic acid anticoagulant was used.
Methods
Conventional cytogenetics
Conventional cytogenetics and FISH were performed at the Division for Cytogenetics, University Hospital Centre, Zagreb. Conventional cytogenetics was performed using the standard Giemsa banding method. Analysis was performed on bone marrow cells after short-term culture (24 h). The cells were treated with a colchicine and hypotonic solution then fixed and washed in methanol-acetic acid (3:1). After that, the cells were resuspended in fixative and dropped onto slides [8]. At least 20 metaphases per sample were analyzed and captured using a fluorescence light microscope (Carl Zeiss Microlmaging GmbH, Gottingen, Germany), and image analysis was performed using the Ikaros software package (MetaSystems, Heidelberg, Germany). Karyotypes were described according to the criteria of the International System for Human Cytogenetic Nomenclature (ISCN 2009, ISCN 2013) [910].
FISH
The FISH procedure was performed on a fixed cell suspension obtained with a conventional cytogenetic method on microscope slides [8]. A mixture of probe, buffer, and water was applied onto the fixed BM cells according to the manufacturer's instructions. The processes of denaturation (1 min at 73℃) and hybridization (24 h at 37℃) were performed using an automated machine (HYBrite, Abbott Laboratories, IL, USA). Non-specifically bound probe was washed in a water bath for 2 min with special detergent solutions. After the microscope slide was allowed to air dry, it was treated with the DNA-intercalating dye 4,6-diamidino-2-phenylindole (DAPI) and a glass cover slip was applied.
The Vysis LSI BCR/ABL dual-color dual-fusion translocation test (Abbott Laboratories) was used as a probe since January 2013. This probe is a mixture of the LSI BCR probe labeled with SpectrumGreen and the LSI ABL1 probe labeled with SpectrumOrange. The BCR probe labeled with SpectrumGreen that spans a genomic distance of about 1.5 megabases. It begins within the variable segment of the immunoglobulin light chain locus, which extends along chromosome 22 through the BCR gene and ends at a point approximately 900 kilobases (kb) telomeric of BCR. A region of about 300 kb that contains low-copy number repeats was eliminated from the probe, which introduced a gap in the coverage of the probe target. The ABL1 probe labeled with SpectrumOrange comprises 650 kb of sequence, and it extends from an area centromeric of the argininosuccinate synthetase gene (ASS) to telomeric of the last ABL1 exon [11].
A sample of 200 interphase cells for each patient was analyzed using a fluorescence light microscope (Carl Zeiss Microlmaging GmbH, Gottingen, Germany) under ×1,000 magnification with the use of an immersion oil and optical filters (DAPI, red, green, and triplet- a filter that can see DAPI, red, and green signals simultaneously). The images were processed using the ISIS computer program (ISIS software, Altlussheim, Germany). The results were expressed as the percentage of Ph-positive interphase cells per hundred counts.
The cutoff value was determined with the FISH method in 20 patients with a diagnosis unrelated to CML and it was 1%. If the proportion of BCR-ABL1-positive interphase cells exceeded 1% of all cells, the patient was considered positive for the BCR-ABL1 gene rearrangement.
According to established recommendations, the BM samples for cytogenetic analysis and FISH were obtained at diagnosis, 3 months later, and then every 6 months until the achievement of CCyR [12].
After that, they were analyzed every 12 months. Despite the recommendation, the sample was sometimes sent only for RQ-PCR and therefore a smaller number of cases were monitored for CCyR than for MMR.
RQ-PCR
RQ-PCR was performed at the Laboratory for Molecular Hematology, University Hospital Centre, Zagreb standardized for reporting results according to the International Scale (IS). For each patient, duplicate samples of 2×107 PB leukocytes were homogenized with TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA), followed by the extraction of RNA. The reverse transcription of 2 µg RNA was performed using the high-capacity cDNA reverse transcription kit with RNase inhibitor (Thermo Fisher Scientific). The amplifications of the fusion gene BCR-ABL1 in addition to ABL1 as a control gene were performed on a LightCycler 2.0 (Roche, Basel, Switzerland) using an ipsogen BCR-ABL1 Mbcr Kit (Qiagen, Venlo, The Netherlands), according to the manufacturer's instructions. All the samples were analyzed in duplicate for both BCR-ABL1 and ABL1. The results were calculated using a Mannheim-derived conversion factor according to laboratory recommendations as BCR-ABL1/ABL1% IS [1314]. RQ-PCR was performed on PB samples soon after diagnosis and 3, 6, 9, and 12 months later [12].
Statistical analysis
Categorical data was compared using Fisher's exact test. Time to CCyR and time to MMR were calculated from the beginning of the treatment and the next 12 months using the Kaplan-Meier method and described with cumulative incidence curves. P-values <0.05 were considered statistically significant. The analysis of statistical data was performed using the software programs MedCalc (MedCalc Software, Ostend, Belgium) and Wolfram Mathematica (Wolfram Research, Champaign, IL, USA).
Go to :
RESULTS
Among 122 patients with positive BCR-ABL1 rearrangement, the median proportion of BCR-ABL1 copies in interphase nuclei was 85% (range, 2–97). Ninety-two (75%) patients showed reciprocal t(9;22) (q34;q11.2) and 30 (25%) showed atypical FISH signals.
All patients in this study started with imatinib mesylate therapy at the dose of 400 mg/day. If their response to therapy was not sufficient, they were switched to nilotinib or dasatinib.
The incidence of atypical patterns in cytogenetic and FISH analyses
Cytogenetic analysis
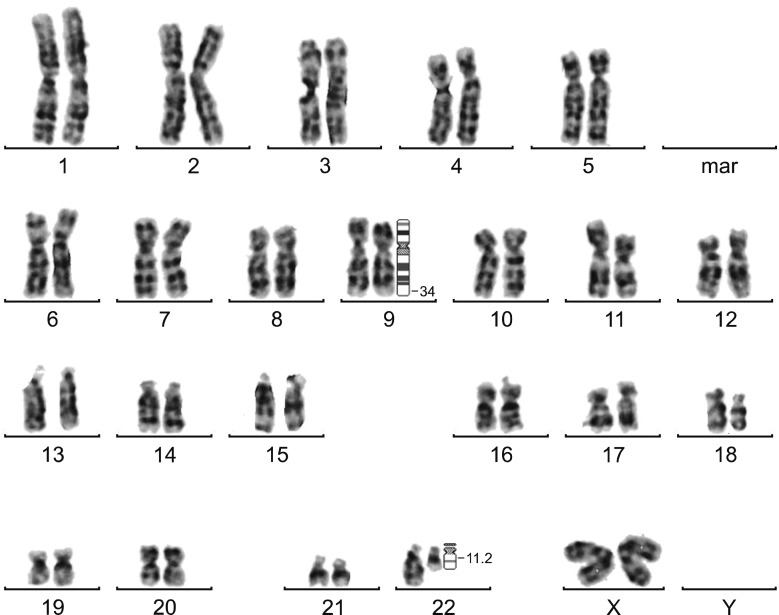
In this study, 70 (57%) samples were evaluated using conventional cytogenetics. Four (40%) of ten cases of variant t(9;22) showed the Ph chromosome on karyogram analysis and six cases (60%) did not show the Ph chromosome due to a lack of cells undergoing cell division. Newly diagnosed CML patients typically have a high level of white blood cells, resulting in a dense BM sample and consequently a low quality of metaphases, which can impede the success of karyotyping.
The karyogram of case number (No.) 2 showed t(9;22) with chromosome 1 involved in the translocation, resulting in t(1;9;22)(p35;q34;q11.2). The karyogram of case no. 10 showed that the third chromosome involved in t(9;22) was chromosome 9, resulting in t(9;9;22)(q34;q34:q11.2) (Fig. 1). Two cases (No. 1 and No. 4) only showed t(9;22) and no involvement of the third chromosome was detected. The submicroscopic deletion of del/der(9) cannot be detected by conventional cytogenetic methods and karyotype analysis.
FISH analysis
All the samples were successfully analyzed with FISH. Generally, in negative interphase nuclei for t(9;22), the signal pattern consisted of two red signals (two normal copies of chromosome 9) and two green signals (two normal copies of chromosome 22), which was termed 2R2G (Fig. 2A). With the dual-color dual-fusion BCR/ABL probe, the positive reciprocal t(9;22) signal pattern revealed two fusion signals (derivative chromosome 9 and derivative chromosome 22), one red signal (normal chromosome 9), and one green signal (normal chromosome 22), which was termed 2F1R1G (Fig. 2B). Cases with an atypical FISH signal pattern comprising one fusion signal (derivative chromosome 22), two red signals (normal and derivative chromosome 9), and two green signals (normal chromosome 22 and a third involved chromosome) were termed 1F2R2G (Fig. 2C) and represent variant t(9;22). We noted variant t(9;22) in 10 (8.2%) of the samples (5 females and 5 males). Their median age was 49 years (range, 29–69). The median proportion of BCR-ABL1 copies in interphase nuclei was 79% (58–90). All 10 CML cases with a variant translocation are described in Table 1.
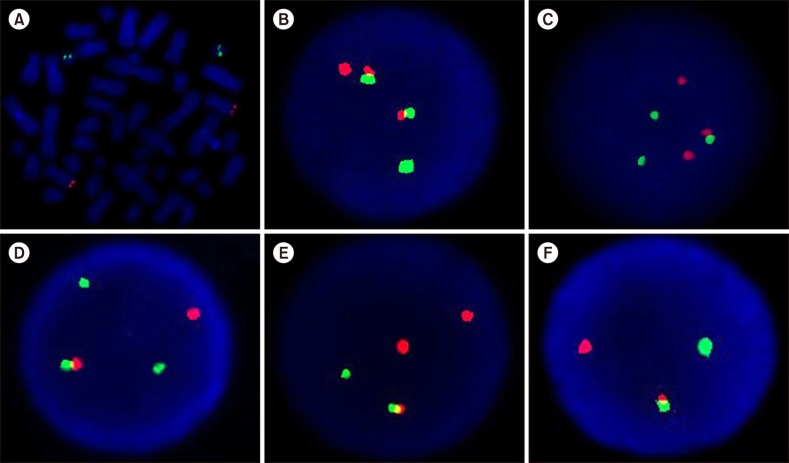 | Fig. 2Representative FISH signal patterns using LSI BCR/ABL dual color dual fusion probe. (A) Metaphase cell, negative, 2R2G, (B) interphase nuclei carrying positive reciprocal t (9;22), 2F1R1G, (C) variant t (9;22), 1F2R2G, (D) ABL1 deletion, 1F1R2G, (E) BCR deletion, 1F2R1G, (F) ABL1-BCR deletion, 1F1R1G.
|
The next three atypical FISH signals described represented deletions. Ten (8.2%) samples of reciprocal t(9;22) with additional cytogenetic abnormalities such as an ABL1 deletion had one fusion signal (derivative chromosome 9), one red signal (normal chromosome 9), and two green signals (one from normal chromosome 22 and one from reciprocally translocated chromosomes 9 and 22) and were termed 1F1R2G (Fig. 2D). The calculated median of BCR-ABL1 copies in those cases was 86% (range, 15–93).
Four (3.3%) samples with a BCR deletion revealed one fusion signal (derivative chromosome 22), two red signals (one from reciprocally translocated chromosomes 9 and 22 and one from normal chromosome 9) and one green signal (normal chromosome 22) and were termed 1F2R1G (Fig. 2E). The median value of BCR-ABL1 copies was 77% (range, 63–89). Six (4.9%) samples with an ABL1 and BCR fusion deletion showed one fusion signal (derivative chromosome 22), one red signal (normal chromosome 9), and one green signal (normal chromosome 22) and were termed 1F1R1G (Fig. 2F). The median proportion of BCR-ABL1 copies was 85% (range, 56–92). Table 2 shows the details of 20 CML cases with del/der(9).
Cytogenetic response
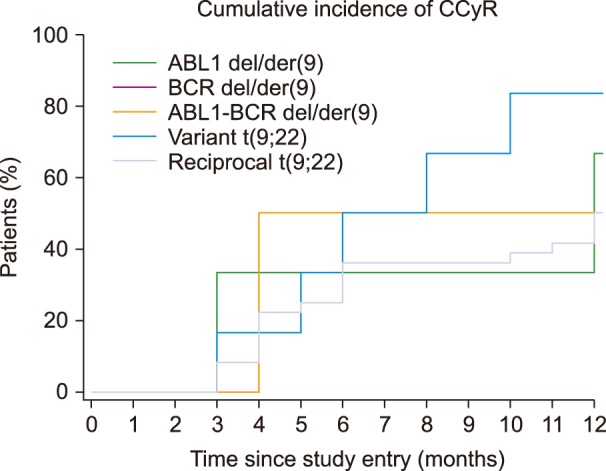
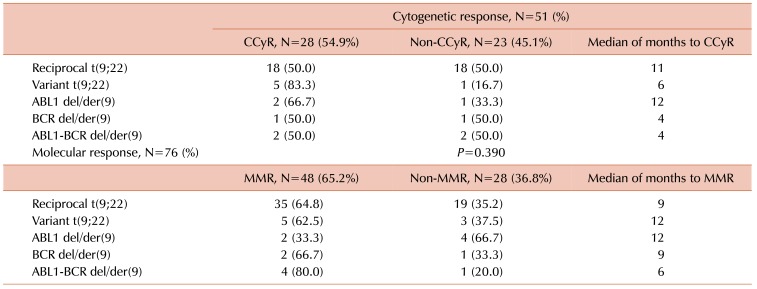
Of the 122 cases with a diagnosis of CML, 51 were included in our database for tracking the cytogenetic response. CCyR was identified in 0% of Ph-positive interphase nuclei on FISH [15]. The results for the cytogenetic response are shown in Table 3 and Fig. 3 overall, 28 (54.9%) patients achieved CCyR within 12 months of starting the treatment, while 23 (45.1%) had not responded to therapy 12 months after starting the treatment.
Molecular response
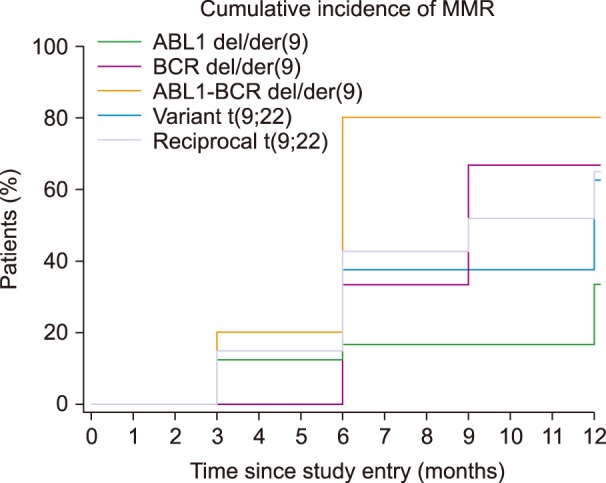
For MMR, 76 of the 122 patients were monitored. MMR was defined as a BCR-ABL1/ABL1 ratio <0.10% on the IS (14). The results for the molecular response are shown in Table 3 and Fig. 4. Within 12 months of starting the treatment, 48 (63.2%) patients achieved MMR while 28 (36.8%) had not responded to therapy 12 months after starting the treatment. The median number of ABL1 copies among all the samples was 194,750 copies (range, 26,000–772,000).
Go to :
DISCUSSION
To our knowledge, this is the first study that evaluated the incidence and molecular-cytogenetic response to TKI of variant t(9;22) and del/der(9) in Croatian CML cases. In the present study, variant translocation occurred in 8.2% of patients, similar to results of Marzocchi et al. who found 5–10% of newly diagnosed CML cases with a variant translocation. The mechanisms of the variant translocations are not fully clear, but Marzocchi et al. [16] have suggested 1-step and 2-step mechanisms (Fig. 5).
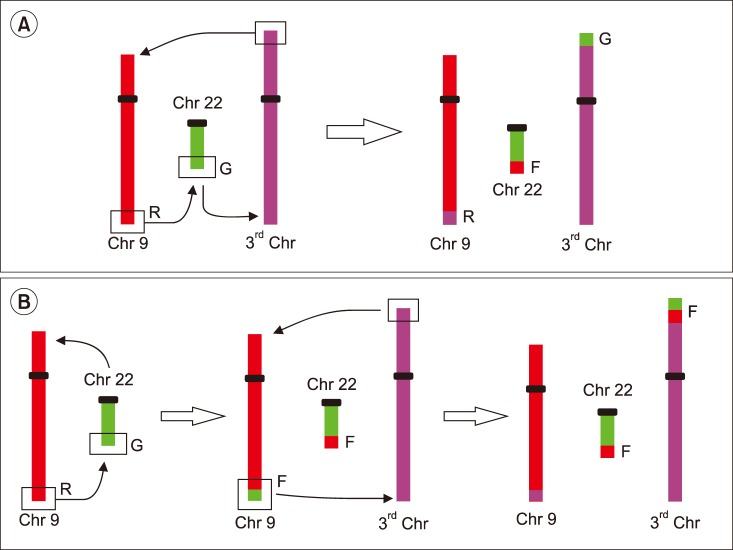 | Fig. 5Mechanisms of variant translocations. (A) 1-step mechanism in which chromosome breakage occurs simultaneously on 3 or 4 different chromosomes in 3-way or 4-way translocation and (B) 2-step mechanism involving 2 sequential translocations in which a reciprocal t(9;22) is followed by a second translocation involving additional chromosomes.
|
Nevertheless, 83.3% patients with variant t(9;22) achieved CCyR and 62.5% achieved MMR. In our study, the presence of variant translocation did not have an influence on CCyR or MMR, which were achieved within a year. Compared to reciprocal t(9;22) patients, 50.0% of them reached CCyR and 64.8% reached MMR. For all patients with variant translocation, response durations were similar to those of patients with reciprocal Ph translocation.
Significance of del/der(9) in patients on TKI therapy is still uncertain, but a number of studies showed that this deletion occurs at the same time as the formation of Ph chromosome [1718192021]. Sinclair et al. [17] and Huntly et al. [18] suggested a significant association between the presence of der/del(9) and a poor prognosis, but yet only for patients who have received interferon. For this reason, the European LeukemiaNet recommendations considered der/del(9) as a candidate for adverse prognostic factor in 2006, which requires a careful monitoring of patients on TKI [22]. Present study showed 16.4% of cases with del/der(9), similar to published results of 10–15% patients with CML [19].
In this study, data showed that 4.9% of patients had ABL1-BCR deletion. Lim et al. [7] demonstrated the presence of such a pattern in about 10.4%. Ability of lacking ABL1-BCR expression suggests that there is a mechanism by which ABL1-BCR transcription can be abolished. Huntly et al. reported 65% of patients who lack ABL1-BCR expression, do not have deletion that is detectable by FISH method. It is likely that small deletions appear to abolish ABL1-BCR transcription, but that would be below the threshold of detection for dual-fusion FISH technique as it uses a large probe [2023]. This study showed 50.0% of patients with ABL1-BCR deletion achieved CCyR, and 80.0% achieved MMR within 12 months. Compared to reciprocal t(9;22) patients, 50.0% of them reached CCyR and 64.8% reached MMR. The significance of ABL1-BCR expression in CML and existence of stable fusion protein, have not been fully clarified [182024]. Huntley et al. [20] showed that the loss of ABL1-BCR transcription is not associated with reduced survival or reduced length of the chronic phase of CML. This demonstrates that lack of ABL1-BCR expression is not sufficient for the poor prognosis associated with overt del/der(9).
Deletion of BCR region was found in 3.3% patients, which was comparable to 2.1% and 2.5% found by Lim et al. [7] and Jain et al. [19]. The function of the normal BCR gene is not clear. It is known that the BCR region that translocates to del/der(9) encodes a GTP-ase-activating protein for p21rac, which affects cell growth and proliferation. GTP-ase activating protein inhibits the activity of p21rac by binding to it. There is the possibility that loss of this region can induce abnormal cell growth and proliferation. Many tumor-related genes are located near the translocation breakpoint, which can also be a reason for potential poor prognosis [24]. Southern blot analysis identified small deletions on chromosome 22, adjacent to the Ph chromosome translocation breakpoint, but without the pathophysiological importance [17]. In our results, 50.0% of patients with deletion of BCR achieved CCyR, and 66.7% MMR within 12 months. Considering the results of CCyR and MMR of patients with reciprocal t(9;22), there is no difference between the patients with and without deletion of BCR region. González et al. [25] reported a lack of difference between the group with the deletion and the group without deletion, which supports the hypothesis that the ABL1-BCR fusion gene containing the major BCR (M-BCR) sequence 3′ to the breakpoint does not play the main role in the leukemogenesis of CML.
Morel et al. [21] reported 9% and Lee et al. [24] demonstrated 7% patients with ABL1 deletion only and these were comparable to 8.2% found in present study. CCyR and MMR were achieved in 66.7% and 33.3% patients with deletion of ABL1, respectively. In several studies, it was established that some CML patients had large deletions adjacent to the ABL1 breakpoint [7172126] resulting in the loss of one or more tumor suppressor genes [17]. Cohen et al. [26] studied the differences of the gene expression profiles between the deletion and non-deletion 9q patients using the cDNA microarray technology. Their studies revealed a number of genes with modified expression patterns, many of which play a role in cell migration or cell-cell/cell-matrix interactions. Proteins such as matrix metalloproteinases (MMPs) and intercellular adhesion molecule-1 (ICAM-1) play an important role in cell migration, whereas proteins such as migration inhibitory factor related protein (MRP), alpha-1 protease inhibitor and neutrophil elastase inhibitor play an important role in inhibition of chemotaxis and random migration. It has been proposed that the invasion, digestion of extracellular matrix, and chemotaxis of cells are mediated by the correct balance between extracellular matrix proteases, the surrounding antiproteases, and the presence of adhesion molecules on the cell surface. Abnormalities of this balance in either direction could lead to a cancerous transformation [26]. The time at which the deletion occurs and the prognostic implications of the 5′ ABL1 region deletion on the der(9) have not as yet been elucidated and their answer remains controversial [21].
The only difference in achievement of MMR was observed for deletion of ABL1 on derived 9 chromosome where only 33.3% of patients with ABL1 deletion achieved MMR in contrast to more than 60% in other groups. Despite this percentage, Kaplan-Meier statistical analysis showed P=0.390. However, it should be taken into account that this study involved a small number of patients and a short monitoring time.
This study showed that the percentage of variant translocation and del/der(9) in Croatian population is in agreement with the data from previously published studies from around the world. A number of studies agree that atypical patterns have no impact on the prognosis of patients with CML [220]. In our series of patients, it was not conclusive that a significant difference in achievement of CCyR and MMR exists between reciprocal translocation and variant translocation, deletion of ABL1, BCR, and ABL1-BCR fusion gene.
Go to :
 XML Download
XML Download