

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
According to a World Health Organization (WHO) report, β-thalassemia and sickle cell disease (SCD) are the world's most common hereditary hemoglobinopathies and approximately 5% of the world's population carries trait genes for these hemoglobin disorders [1]. Beta-thalassemia shows a high prevalence in the Mediterranean, Middle Eastern, and South Asian regions, while SCD is endemic to Central Africa. However, population migration has resulted in hereditary hemoglobinopathies becoming a global and growing health problem in many countries [23].
Therapy for thalassemia major (TM) includes supportive treatment such as transfusion and iron chelation therapy. However, allogeneic hematopoietic stem cell transplantation (HSCT) remains the only curative treatment option. Similarly, allogeneic HSCT is curative for SCD, although hydroxyurea may reduce morbidity and mortality by increasing fetal hemoglobin and decreasing red blood cell (RBC) sickling. The timing of HSCT in SCD, however, is controversial, with HSCT often recommended when patients begin to experience SCD-related complications such as stroke, acute chest syndrome, and recurrent vaso-occlusive pain crisis.
TM and SCD are both caused by genetic defects in the hematopoietic system, and may be cured by gene therapy, although such a treatment modality is not available in most clinical settings [456].
Patients with severe thalassemia and sickle cell anemia requiring HSCT have not yet been reported in Korea, although the incidence of severe hemoglobinopathy is expected to rise in the future [3]. In this study, we report the outcome of TM and SCD patients who received allogeneic HSCT with myeloablative conditioning at our institution, with the aim to present baseline results that may be utilized to improve outcome in the future.
Go to : 
MATERIALS AND METHODS
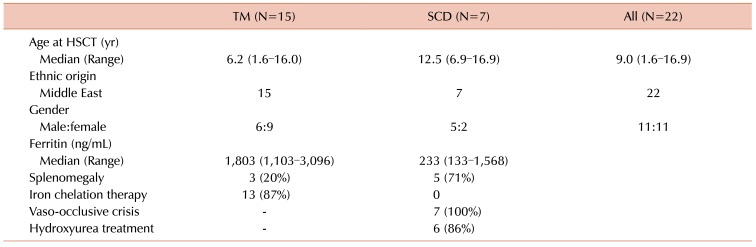
Patients
We retrospectively reviewed data on 22 consecutive TM or SCD patients who received allogeneic HSCT from July 2012 to December 2015 at the Department of Pediatrics, Catholic Blood Hospital, The Catholic University of Korea. This study received approval from the institutional review board (IRB). The study group included 15 patients with TM and 7 patients with SCD (11 males, 11 females) (Table 1). The median age at transplantation was 9.0 years (range, 1.6–16.9). All patients were of Middle Eastern ethnicity. The median ferritin level of TM patients was 1,803 ng/mL (range, 1,103–3,096), and 13 patients received iron chelation therapy. All SCD patients had previously experienced vaso-occlusive crisis, and received hydroxyurea therapy at the time of HSCT, except for 1 patient, intolerant to the medication.
Donor and conditioning regimen
All donor–recipient pairs were matched according to high resolution typing for HLA-A, B, C, and DRB1 alleles. Donor types in the HSCTs were as follows: 18 matched sibling donors (MSD), 3 mismatched related donors (including 2 haploidentical and 1 HLA-DR 1-antigen mismatched donor), and 1 matched unrelated donor (MUD). Although 12 donors had β-thalassemia trait and 3 donors had sickle cell trait, none of the donors showed evidence of hemoglobinopathy (Table 2).
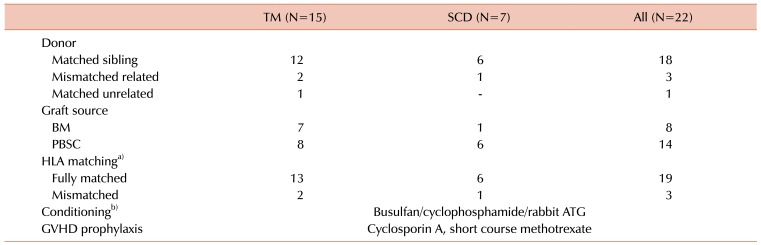
Table 2
Transplantation characteristics.
a)HLA-A, -B, -C, -DR matching with DNA typing. b)With the exception of one haploidentical transplantation HSCT, conditioning was performed with total body irradiation, busulfan, fludarabine and ATG.
Abbreviations: ATG, anti-thymocyte globulin; BM, bone marrow; GVHD, graft-versus-host disease; HLA, human leukocyte antigen; PBSC, peripheral blood stem cell; SCD, sickle cell disease; TM, thalassemia major.
![]()
Twenty patients received the following conditioning regimen: intravenous (IV) busulfan 130 mg/m2 once daily for 4 days, cyclophosphamide 60 mg/kg once daily for 2 days and anti-thymocyte globulin (ATG) (Thymoglobulin; Genzyme, Cambridge, MA, USA) 2.5 mg/kg once daily for 3 days. One patient received the same conditioning regimen except that cyclophosphamide was administered at 50 mg/kg for 4 days. One SCD patient, who underwent haploidentical HSCT, received total body irradiation 400 cGy, busulfan 130 mg/m2 once daily for 2 days, fludarabine 30 mg/m2 for 5 days and ATG 2.5 mg/kg for 3 days. The other TM patient received haploidentical HSCT from a homozygous HLA donor with 4/8 match in the graft-versus-host direction, and 8/8 match in the host-versus-graft direction; this patient received the same conditioning as MSD HSCT patients.
Transplantation care
Graft-versus-host disease (GVHD) prophylaxis consisted of IV cyclosporine (3 mg/kg/day IV initially, followed by 5 mg/kg/day oral dose, when oral intake was possible) starting from day −1 and 4 infusions of short course methotrexate (5 mg/m2) at days 1, 3, 6, 11. In the absence of GVHD, cyclosporine was maintained at therapeutic levels (100–200ng/mL) until 9 months post-transplantation, then subsequently tapered and stopped at 1 year post-transplantation. Antimicrobial prophylaxis included oral acyclovir from the beginning of conditioning until day 42, and oral trimethoprim-sulfamethoxazole from the beginning of conditioning until day −3, and from neutrophil engraftment until at least 1 year after transplantation. Antifungal prophylaxis consisted of IV micafungin (1 mg/kg/day) from the beginning of conditioning until neutrophil engraftment, at which point oral fluconazole was administered and maintained for at least 6 weeks. G-CSF was administered from day 5 until absolute neutrophil count (ANC) ≥3.0×109/L.
Detection of post-HSCT cytomegalovirus (CMV) DNAemia and Epstein-Barr virus (EBV) DNAemia was performed with real-time quantitative monitoring of plasma samples after engraftment at weekly and fortnightly intervals, respectively, until 3 months after transplantation, and at longer intervals subsequently. Preemptive treatment for EBV DNAemia was not provided, and rituximab was only administered with diagnosis of post-transplantation lymphoproliferative disease (PTLD).
Immune reconstitution and chimerism analyses were performed at 1, 3, 6, 9 and 12 months post-HSCT. Chimerism was determined with short tandem repeat polymerase chain reaction (STR-PCR) analysis of peripheral blood, with full donor chimerism defined as >95% donor hematopoietic cells, and mixed donor chimerism defined as 5–95% donor-derived cells.
Definition and evaluation of data
Date of neutrophil engraftment was defined as the first of 3 consecutive days with ANC ≥0.5×109/L, and the date of platelet engraftment was defined as the first of 3 consecutive days with platelet count ≥50×109/L without platelet transfusions in the preceding week. Acute GVHD was graded according to published consensus criteria, and chronic GVHD was classified according to revised 2014 NIH criteria [78]. Major toxicities were graded according to NCI CTCAE (version 4.0). Hepatic veno-occlusive disease (VOD) was diagnosed according to clinical criteria as the presence of two of the following: 1) hyperbilirubinemia (bilirubin >2 mg/dL), 2) painful hepatomegaly, and 3) unexplained weight gain (>5% from baseline).
Statistical analysis
Primary outcomes analyzed were the incidences of graft failure and treatment-related mortality (TRM) after HSCT. Secondary endpoints included the incidence of VOD and acute and chronic GVHD. Overall survival (OS) was calculated using the Kaplan–Meier method. Both acute and chronic GVHD were calculated using the cumulative incidence function, with TRM without respective GVHD as a competing risk [9]. Patients alive were censored at date of last follow-up, but no later than April 30, 2017. Statistical analyses were performed with R package version 2.14.2 (R Foundation for Statistical Computing, Vienna, Austria).
Go to :
RESULTS
Engraftment and early complications
Graft failure was not observed, and all patients showed neutrophil and platelet engraftment at a median time of 14.5 days (range, 8–25), and 12.5 days (range, 8–65), respectively. Median donor chimerism at 1 month post-transplantation was 99% (range, 93–100) (Table 3). Twenty-one out of 22 patients showed complete donor chimerism at this time point. During the last follow-up, 2 MSD bone marrow transplantation (BMT) recipients, who initially achieved complete donor chimerism, showed a decrease in chimerism down to 94% and 81%, while 1 patient with initial partial donor chimerism converted to complete donor chimerism. All TM patients became transfusion-independent with normal complete blood counts.
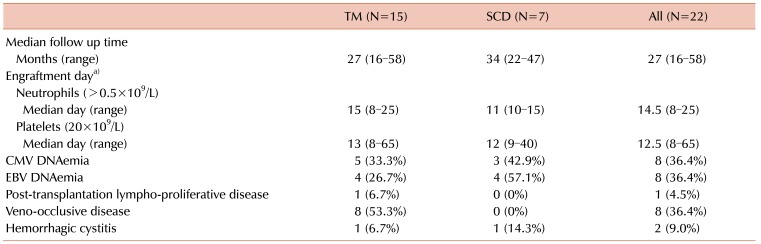
As TM and SCD are at high risk for VOD, and as 1 of the 2 initial patients showed VOD, prophylaxis against VOD was started for the third HSCT patient with alprostadil (Eglandin, Welfide Korea, Hwaseong, Korea) at a dose of 0.1 µg/kg/day with continuous infusion starting 1 day prior to conditioning until 30 days after HSCT. No significant difference in VOD incidence was observed according to median ferritin levels: 36.4% [95% confidence interval (CI), 21.9–50.9] for lower ferritin levels and 45.5% (95% CI, 30.5–60.5) for higher ferritin levels (P=0.636). VOD occurred in 8 out of 15 patients with TM (53.3%) and in none of the SCD patients (Table 3). Severity of VOD was as follows: 1 mild, and 7 moderate. Median time to VOD onset was 13 days (range, 9–26), and median highest total bilirubin was 3.9 mg/dL (range, 1.7–11.2). All 7 patients with moderate VOD showed abdominal pain, and disseminated intravascular coagulopathy requiring antithrombin III treatment. Median weight gain percentage was 15% (range, 6.5–27.5). All VOD patients received either recombinant tissue plasminogen activator (TPA) or defibrotide as anticoagulant therapy, with 12.5 days median duration of treatment (range, 9–26). Supportive care also included glutathione and vitamin E treatment. All patients showed resolution of VOD.
Hemorrhagic cystitis occurred in 1 patient in each of the TM and SCD patient groups.
GVHD incidence
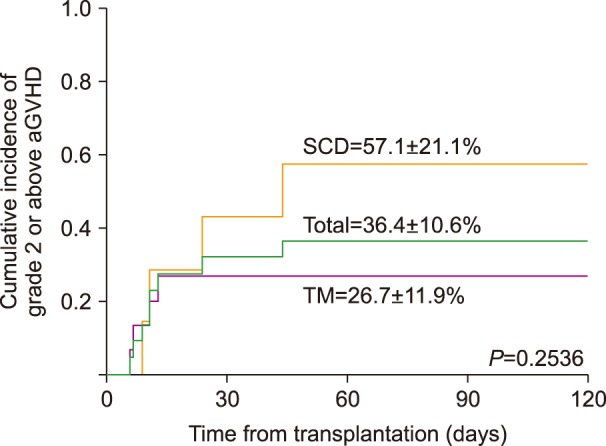
Overall, 8 patients were diagnosed with and treated for acute GVHD of grade 2 or above with a cumulative incidence of 36.4% (95% CI, 25.8–47.0) (Fig. 1). Four of these patients experienced grade 3 or 4 acute GVHD (1 TM and 3 SCD patients) and 1 SCD patient died of complications resulting from grade 4 gut GVHD. PB was the cell source for these 5 patients. Cumulative incidence of acute GVHD of grade 2 or above was higher in the 14 patients who underwent peripheral blood stem cell transplantation (PBSCT, 50.0%, 95% CI, 36.6–63.4), compared with the 8 patients, who received BMT (12.5%, 95% CI, 0.8–24.2), although the number of patients was too low to determine statistical significance (P=0.072). Other factors such as primary disease (TM vs. SCD) and donor type (MSD vs. others) did not significantly influence acute GVHD incidence.
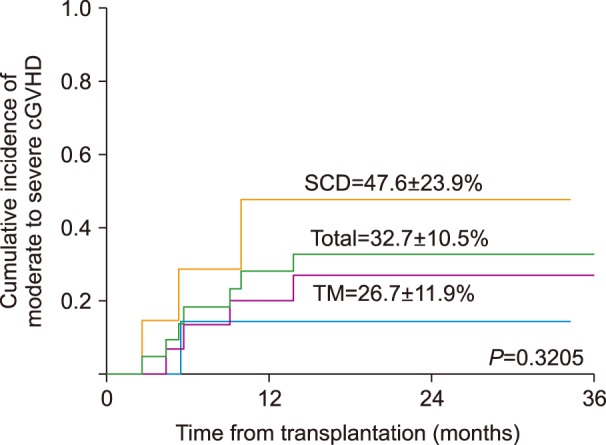
Eight patients were diagnosed with chronic GVHD and the severity of chronic GVHD was as follows: 1 mild, 2 moderate, and 5 severe. One patient with de novo mild chronic GVHD showed oral GVHD and dry eye symptoms, but showed rapid resolution of disease with oral prednisolone administration. The cumulative incidence of moderate to severe chronic GVHD was 32.7% (95% CI, 22.2–43.2) (Fig. 2). The cumulative incidence of chronic GVHD of at least moderate severity was higher for PBSCT recipients compared with BMT recipients, although without statistical significance (38.9%, 95% CI, 25.1–52.7 vs. 25.0%, 95% CI, 9.7–40.3, P=0.467). Of the 7 patients with chronic GVHD of moderate severity or above, 2 were diagnosed with de novo chronic GVHD, while 5 showed preceding acute GVHD. The following organs were involved in chronic GVHD: the lungs (5 patients), the skin (3 patients), the oral cavity (3 patients), and the eyes (2 patients). Three patients had lung GVHD alone, of whom 1 PBSCT recipient died of this complication. Four patients continued to receive immunosuppressive therapy during the last follow-up, while GVHD treatment ceased for 3 patients without disease progression.
Infectious complications
CMV DNAemia was detected in 8 patients (36%), with initial preemptive ganciclovir treatment resulting in resolution of DNAemia in all instances (Table 3). EBV DNAemia was detected in 8 patients and 1 patient with thalassemia progressed to PTLD, which resolved with rituximab therapy.
Survival results
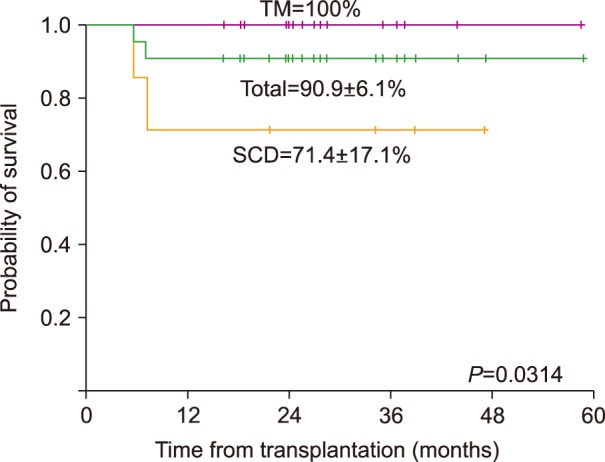
The estimated 2-year OS for the entire cohort was 90.9±6.1%, with a median follow-up of 27 months (range, 16–58) (Fig. 3). All TM patients survived, while 2 patients with SCD died; 1 from sepsis subsequent to grade 4 acute gut GVHD after MSD PBSCT, and another patient from severe chronic lung GVHD after MSD PBSCT. All surviving SCD patients remained free of SCD-related complications, such as vaso-occlusive crisis.
Go to :
DISCUSSION
In thalassemia, extramedullary hematopoiesis leads to bone deformities and hepatosplenomegaly, while regular packed RBC transfusions result in iron overload that requires iron chelation therapy. All but 2 of the TM patients received iron chelation at the time of HSCT; however, serum ferritin remained high due to regular RBC transfusions at 3–4 weekly intervals.
In SCD, hemoglobin S (HbS) polymerization may lead to vaso-occlusive disease and subsequent severe organ damage. Hydroxyurea has been used since the 1990s to minimize both transfusion need and vaso-occlusion [1011]. All 7 SCD patients in our study had previously received hydroxyurea, including 6 who were receiving the medication until two weeks prior to HSCT. Common indications for HSCT in SCD include recurrent vaso-occlusive disease, evidence of central nervous system (CNS) complications, or osteonecrosis. However, improved HSCT outcomes have led to HSCT being considered prior to the onset of disease-related complications [12]. All SCD patients in our study had previously experienced vaso-occlusive disease, leading to HSCT consideration. None of the patients, however, had experienced stroke or other CNS events.
The majority of HSCT donors in our study were MSDs (18/22, 82%), with 12 and 3 patients showing thalassemia or sickle cell trait, respectively. All donors were eligible for HSCT donation after pre-transplantation evaluation.
In contrast to allogeneic HSCT in malignant hematological diseases, normal immune competence and bone marrow hyperactivity, and allo-immunization due to multiple transfusions may result in a high incidence of mixed chimerism and graft rejection for TM or SCD patients receiving HSCT. Hence, myeloablative conditioning is the standard treatment for patients without significant organ dysfunction [13]. Busulfan- and cyclophosphamide-based conditioning regimens are commonly used, with ATG added to prevent graft rejection [141516]. The majority of our patients received such a conditioning regimen, resulting in no graft rejection and transfusion independence in all surviving patients, with all but 2 patients showing complete donor chimerism during the last follow-up. All surviving SCD patients also remained free of SCD-related complications. However, busulfan-based conditioning may increase TRM and VOD incidence, while transfusion-related iron overload is also known as a risk factor for VOD [1718]. In our study, VOD occurred in 8 patients in the TM group (36.4%), resulting in a significantly higher incidence than the average VOD incidence for HSCT [19]. Transfusion-related iron burden and busulfan-based myeloablative conditioning most likely acted as major risk factors. All cases of VOD in our study resolved with therapy including TPA, defibrotide, vitamin E, and glutathione. However, the high incidence of VOD, as well as late effects including gonadal failure, emphasize the need to consider a non-myeloablative conditioning regimen [2021].
Recent studies on HSCT for TM or SCD patients showed that OS was over 90% [1222]. Hence, the current focus is on reducing TRM and long-term toxicities through non-myeloablative or reduced toxicity conditioning, although no one method is considered as the standard treatment [2324]. Our patients live in foreign countries and follow-up is difficult, therefore we used myeloablative conditioning for better engraftment and to improve long-term prognosis. We have also recently utilized a treosulfan-based conditioning (treosulfan, thiotepa, fludarabine, ATG) in 2 patients who did not experience VOD, although additional studies are necessary.
MSD-HSCT in TM or SCD patients results in a 10–20% incidence of acute or chronic GVHD [1222]. However, when PB was used as the cell source, the incidence of GVHD has been reported to increase [25]. The incidence of GVHD in our study was high (36.4% for grades 2–4 acute GVHD, 32.7% for chronic GVHD), and in 6 out of 7 PBSCT SCD patients, the incidence of acute and chronic GVHD was 57.1% and 47.6%, respectively. Although the low number of cases limits statistical significance, the incidence of both acute GVHD (50.0% vs. 12.5%) and chronic GVHD (38.9% vs. 25.0%) was higher in PBSCT than in BMT, indicating that BM is the optimal cell source for HSCT.
Unrelated donor or haploidentical donor transplants are currently undertaken for patients with hemoglobinopathy, who lack an MSD [262728]. One patient who received an MUD-HSCT did not develop GVHD, although both haploidentical HSCT recipients developed grade 3 acute GVHD and subsequent moderate and severe chronic GVHD. These patients are currently receiving tapered immunosuppressive treatment.
Although our study is limited by the low number of patients treated at a single pediatric institution in Korea with short-term follow-up, we found that treatment of patients with congenital hemoglobinopathy with busulfan/cyclophosphamide/ATG-based myeloablative conditioning resulted in engraftment and transfusion independence in all surviving patients. However, the rate of VOD was high and 2 patients died of GVHD, despite receiving HSCT from an MSD.
Reduced intensity conditioning (RIC) may be considered to reduce TRM in hemoglobinopathy patients who receive HSCT. A recent study of 52 hemoglobinopathy patients treated with RIC comprised of fludarabine, melphalan and alemtuzumab followed by either BMT or cord blood transplantation reported 92.3% event-free survival, 5.7% TRM, and acute and chronic GVHD incidences of 23% and 13%, respectively [29]. Although the incidence of mixed chimerism was high (27%), all patients remained transfusion-independent and SCD patients remained free of vaso-occlusive complications.
For patients who lack an MSD or MUD, haploidentical donor HSCT may be considered. The key issue in haploidentical donor HSCT is the potential for severe GVHD, which may be prevented by methods such as post-transplantation cyclophosphamide administration or ex vivo depletion of T cells from the graft. Recent reports of haploidentical donor HSCT in hemoglobinopathy patients showed limited rates of serious toxicities, although the high rate of graft rejection remained a significant problem [272830].
In summary, we report out results from myeloablative conditioning-based HSCT for pediatric hemoglobinopathy patients. Although survival outcomes were acceptable, additional methods to ease early HSCT complications and reduce GVHD incidence are necessary.
Go to :
 XML Download
XML Download