

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
BIS (BCL-2 interacting cell death suppressor), also called BAG3, has been originally identified as an anti-apoptotic protein and cochaperone which binds to BCL-2 and HSP (heat shock protein)70, respectively [12]. Evidence subsequently showed that BIS was involved in diverse cellular functions including anti-apoptotic, anti-stress, migration and invasion, autophagy, development, and cell senescence, which might be mediated by its various binding partner proteins [3]. Clinically, BIS has been implicated in several pathological states including human cancers of various origins, which was supported by its pro-survival function [45]. BIS has been also studied in myopathy and neurodegeneration, which is related with its protein quality control ability, targeting z-disk composing proteins or aggregation-prone proteins [67891011]. Therefore, the regulation of BIS expression might be critical for cellular resistance to various stresses or the maintenance of integrity of cytoskeletal organization, as well as protein homeostasis under physiological and pathological conditions.
While BIS is expressed constitutively at high levels in skeletal and cardiac muscles as well as in various human cancers, BIS has been reported to be induced by stress such as heat shock, heavy metals, serum deprivation, electrophile stress, and oxidative stress [12]. Moreover, BIS expression is considerably elevated upon proteasome inhibition, viral infection, as well as in response to fibroblast growth factor 2 (FGF-2) [131415]. Recently, the 3′-UTR of BIS mRNA was shown to be the target for microRNA (MiR)-371a-5p, MiR143, or MiR-217-5p, resulting in upregulation of BIS in cardiomyocytes and downregulation in glioblastoma and colorectal cells, respectively [161718]. The expression of BIS, however, is mainly regulated at the transcriptional level by activation of several transcription factors [192021]. Among those, heat shock factor 1(HSF1) is the representative transcription factor that regulates BIS expression by interacting with heat shock-responsive elements (HSEs) located in the promoter region of BIS, similar to other stress-responsive genes such as HSP70 [22232425]. Furthermore, ectopic expression of BIS was shown to activate its own promoter as well as HSP70, through a positive feedback loop involving HSF1 [2627].
In addition to a main transcription regulator of BIS expression, HSF1 was recently identified as a binding protein that binds to BIS protein, as demonstrated by a BIS interactom analysis based on quantitative immunoprecipitation combined with knockdown (QUICK) and human proteome microarrays [28]. Recent evidences suggest that HSF1 is constitutively active in cancer cells in contrast to the rapid inactivation upon removal of stress in physiological state, but the molecular mechanism for its persistent activation in cancers is not poorly understood [2930]. Therefore, it is possible that interaction of HSF1 and BIS might modulate the protein stability or localization of HSF1, thereby resulting in accumulation of HSP70 and HSP27 which might confer tumor cells resistant against proteotoxic stress and consequent expansion of tumor cells. However, the significance or requirement of the interaction of HSF1 and BIS on their respective functions is not clearly defined yet.
Therefore, the present study is designed to investigate the consequence of BIS depletion on the HSF1 activity as a transcriptional factor using BIS depletion strategy. Our results showed that activation of HSF1 by several stresses was not affected by BIS knockout or knockdown as determined by the endogenous HSP70 and HSP27 mRNAs as well as HSP70 promoter activity. However, the expression of the BIS gene that also contains HSEs in its promoter region was reduced specifically in the BIS knockout (KO) cells. Our results suggested that BIS was not critically required for HSF1 activity, but was required for its own expression, which involved a HSF1-independent pathway.
METHODS
Cells and treatment
A549 human lung cancer cells and U87 glioblastoma cells were obtained from American Type Culture collection and maintained in DMEM supplemented with 10% feat-inactivated fetal bovine serum (Biowest, Nuaillé, France). The complete depletion of BIS protein was achieved by CRISPR/Cas9 system as previously described [31]. DNA sequencing revealed that only single type of deletion (14 bp) was found in BIS-knockout (KO) clone used in this study while mixed genotypes were observed in the BIS clone used in previous study [31]. Transient knockdown of BIS or HSF1 expression was performed by transfection of 200 nM of specific small interfering RNA (siRNA) for BIS (5′-AAGGUUCAGACCAUCUUGGAA-3′) or HSF1 (5′-CUGAAGAGUGAAGACAUAAAGA-3′) using G-fectin (Genolution, Seoul, Korea). To induce stress response, cells were exposed to heat shock at 43℃ for 30 min and further incubated at 37℃ for recovery as indicated. Cells were also treated with MG132 (Sigma-aldrich, St. Louis, MO, USA), a proteasome inhibitor, as proteotoxic stress. As an oxidative stress, cells were exposed to glucose free and serum free condition for 3 h and then and incubated in the normal media for 3 h. For the determination of BIS mRNA stability, 200 ng/ml of actinomycin D (Sigma-aldrich) was added to cells for indicated times prior to the quantification of BIS mRNA.
Quantitative real time PCR (qRT-PCR)
For gene expression analyses, total RNA was isolated with an RNA extraction kit AcuZol (Bioneer, Daejeon, Korea) and cDNA was prepared with AccuPower® Customized RocketScript Cycle RT premix (Bioneer) according to the manufacturer's protocol. To determine BIS pre-mRNA levels, DNase I (Thermo scientific, Waltham, MA, USA) was treated to exclude the possible contamination of genomic DNA and the reverse transcription was performed using random priming reaction with AccuPower® RocketScript Cycle RT premix (Bioneer). Then, quantitative real-time PCR (qRT-PCR) was performed using SYBR premix Ex Taq (Takara Biotechnology, Shiga, Japan) with specific primers on CFX96 Connect Real-Time PCR Detection System (Biorad, Hércules, CA, USA). The relative values for BIS, HSP70, HSP27 or HSF1 mRNA were calculated after normalizing the Ct value to β-actin levels from the same sample using the ddCt method. For BIS pre-mRNA, normalization was performed using 18S RNA. The specific primers for each mRNA are listed in Table S1.
Western blotting assay
Cell protein extracts were prepared and western blotting analysis was carried out as previously described following standard procedures [32]. The membranes were then incubated overnight with antiserum against BIS [1], HSP70 (Enzo Life Sciences, Farmingdale, NY, USA), HSP27 (Santa Cruz, Santa Cruz, CA, USA) or HSF1 (Santa Cruz) followed by appropriate secondary antibodies. The immunoreactive proteins were visualized using enhanced chemiluminescence system (ECL Western Blotting Substrate, Promega, Madison, WI, USA).
Promoter assay
The promoter activity of BIS or HSP70 was evaluated using luciferase reporter assay. A series of reporter plasmid for BIS promoter with various lengths was prepared by PCR with corresponding primer sets from human genomic DNA, and subsequent cloning with KpnI and XhoI sites pGL3 basic plasmid (Promega) as previously described [23]. A 1.04 kb fragment of 5′-franking region of human HSP70 DNA containing two heat shock elements was also amplified with specific primers and subcloned into KpnI and XhoI sites of pGL3 basic plasmid. Following knockdown of BIS expression, A549 cells were transfected with 0.25 µg of indicated reporter plasmids with Fugene 6 (Roche Life Science, Basel, Switzerland) for 24 h. Wild type (WT) A549 and BIS-KO A549 cells were transfected with a BIS or HSP70 promoter for 24 h and then exposed to heat shock. The activity reporter was measured using a Dual-Luciferase reporter assay system (Promega). After normalization with renilla activity, the promoter activities of each constructs are presented as fold change of luciferase activities relative to that from pGL3 basic plasmid, which was taken as 1.0.
GFP-BIS plasmid constructs
To construct the open reading frame of WT BIS gene and the corresponding region of BIS gene with 14 bp deletion, PCR was performed with the cDNA from WT A549 and BIS-KO A549 cells as a template, respectively. After verification of the correct sequences, the PCR product was then digested and cloned into XhoI and EcoRI sites of pEGFP-C1 (Promega). After transfection of these constructs for 48 h, total RNA was extracted and cDNA was prepared following treatment of DNase I (Thermo scientific). The GFP mRNA levels from each constructs were determined by PCR amplification and the PCR products were analyzed by agarose electrophoresis.
RESULTS
Depletion of BIS did not affect HSF1-dependent transcriptional activation in A549 cells
HSF1 is known as a main transcriptional regulator for BIS expression under stress [222325]. Recently, HSF1 was also shown to interact with BIS, but the significance of the physical interaction of these proteins on HSF1 activity was not clearly defined. Previously we established BIS-KO A549 cells by the CRISPR/Cas9 system and demonstrated that BIS depletion sensitizes A549 cells to cisplatin via suppressing the stability of MCL-1 [31]. However, the association of BIS depletion on HSF1 activity was not studied using this KO strategy. Therefore, we investigated here if BIS depletion affected the transcriptional activation of HSF1 target genes in response to several stresses. First, we determined the expression profiles of HSP70 and HSP27 mRNA upon heat shock in WT A549 and BIS-KO A549 cells in which 14 bp was deleted in exon 1 of the BIS gene [31]. Quantitative analysis of mRNA indicated that the induction patterns of HSP70 and HSP27 mRNA levels in response to heat shock were not significantly different between BIS-KO A549 and WT A549 cells (Fig. 1A). At 1 h of recovery following heat shock, the HSP70 mRNA levels were increased to approximately 11-fold both in WT A549 and BIS-KO A549 cells compared to those of WT A549 at normal conditions. The induction of HSP27 levels was sustained up to 6 h of recovery after heat shock both in WT A549 and BIS-KO A549 cells with 3-fold and 3.4-fold increases, respectively. Western blots also revealed that the expressions of HSP70 and HSP27 proteins were not significantly affected by BIS depletion upon heat shock (Fig. 1B). Moreover, treatment with MG132, a proteasome inhibitor, or recovery from serum free condition, an oxidative stress [3334], also increased HSP70 and HSP27 mRNA expression in WT A549 cells, which were not significantly repressed in BIS-KO A549 cells (Fig. 1A).
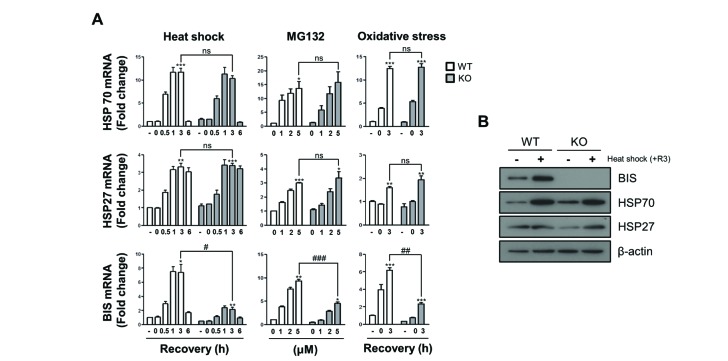
Fig. 1
The induction of HSP70 and HSP27 mRNA was not affected by BIS depletion under stress conditions.
(A) Wild type (WT) A549 cells and BIS-knockout (KO) A549 cells were exposed to heat shock (43℃) for 30 min and subsequent recovery for up to 6 h, to MG132 for 6 h, or to glucose free and serum free conditions for 3 h followed by 3 h of recovery. The mRNA expression levels at the indicated times or indicated treatment concentrations were measured by qRT-PCR analyses. The fold induction was determined as the relative value of each mRNA level compared to the untreated WT A549 cells, which was designated arbitrarily as 1.0. Data are represented as the mean±SEM from three independent experiments. *p≤0.05, **p≤0.01, ***p≤0.001 vs. control in each group; #p≤0.05, ##p≤0.01, ###p≤0.001 vs. WT at the indicated times or concentrations. ns, statistically non-significant. (B) The protein levels of BIS, HSP70, and HSP27 in the WT and BIS-KO A549 were determined by western blotting of samples after 3 h of recovery (+R3) following heat shock and control cell samples. Actin expression was used as a loading control.
![]()
Previously, ectopic expression of BIS was shown to activate its own promoter, involving a 5′-UTR sequence of the BIS gene in glial cells [26]. A subsequent study demonstrated that the positive feedback loop of BIS expression appeared to be a consequence of stress responses generated by the accumulation of BIS protein and subsequently ubiquitination of client proteins, which triggered nuclear translocation of HSF1 [27]. These previous studies prompted us to determine the effects of complete depletion of BIS on the expression of its own mRNA. Because the BIS-KO A549 cells have only the 14 bp deletion in exon 1 of the BIS gene leading to a frame shift, the promoter region harboring several HSEs [2325] should be intact and therefore respond to cellular stresses. As shown in Fig. 1A, the BIS mRNA levels in WT A549 cells were increased by 7.5-fold at 1 h recovery following heat shock while those in the BIS-KO A549 cells were only 2.4-fold increased relative to those in WT A549 cells under non-stressed conditions. In response to proteasome inhibition or oxidative stress, BIS induction was also considerably decreased in BIS-KO A549 cells ; 9.3-fold induction vs. 4.5-fold after treatment with 5 mM of MG132, and 6.2-fold induction vs. 2.3-fold at 3 h recovery after glucose and serum deprivation, respectively. It is noteworthy that, in BIS-KO cells, the basal levels of BIS mRNA were also decreased by 0.45-fold compared to WT A549 cells. These results indicated that BIS depletion inhibited the constitutive as well as inducible expression of the BIS mRNA without alteration of HSP70 and HSP27 mRNA levels.
To exclude the possibility that our results were due to the cellular features acquired during the selection process of BIS-KO A549 cells, which were not relevant with BIS depletion, we transiently suppressed BIS expression in WT A549 cells using siRNA and examined the induction of HSP70 and HSP27 mRNA. Fig. 2A shows that BIS knockdown neither affected the constitutive nor inducible expression of HSP70 and HSP27 mRNAs in response to heat shock, proteasome inhibition, or serum deprivation in conditions in which BIS expression was sufficiently decreased. Thus, these results clearly demonstrated that activation of HSF1 on the HSP27 and HSP70 was not dependent on BIS expression in A549 cells.
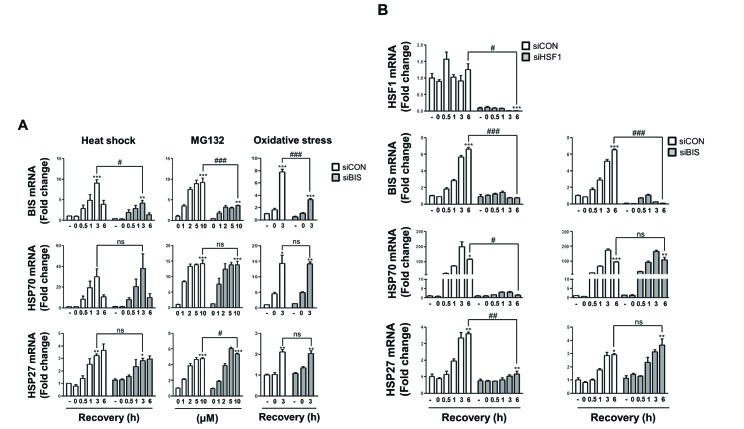
Fig. 2
BIS knockdown did not affect HSP70 and HSP27 induction by various stress conditions.
(A) BIS expression was transiently suppressed by 200 nM of control siRNA (siCON) and BIS specific siRNA (siBIS) for 48 h in A549 cells, and the fold induction was compared for BIS, HSP70, and HSP27 mRNA expression in response to heat shock, MG132, or oxidative stress as described in Fig. 1. (B) U87 cells were transfected with HSF1 siRNA (siHSF1) or BIS siRNA (siBIS) for 48 h and exposed to heat shock. The expression levels of HSF1, BIS, HSP70, and HSP27 mRNA were determined as in Fig. 1. *p≤0.05, **p≤0.01, ***p≤0.001 vs. untreated control cells in each group; #p≤0.05, ##p≤0.01, ###p≤0.001 vs. siCON at the indicated times or treatment concentrations. ns, statistically non-significant.
![]()
Transcriptional activity of HSF1 was not suppressed by siRNA-mediated BIS depletion in U87 glioblastoma cells
Our findings were inconsistent with a previous report showing that BIS knockdown using adenovirus was accompanied by decreases in HSP70 and HSP27 expression as determined by western blots of T98G glioblastoma cells [27]. Thus, to determine if the lack of a functional association of BIS with HSF1 activity was the specific phenotypes of A549 cells, we suppressed HSF1 or BIS expression in U87 glioblastoma cells and examined the HSP70 and HSP27 mRNA levels upon heat shock stress. As shown in Fig. 2B, HSF1 knockdown substantially decreased HSP70 and HSP27 mRNAs. At 6 h of recovery following heat shock, HSP70 and HSP27 mRNA levels were 116-fold and 3.6-fold increased, respectively, compared to unstressed control U87 cells, whereas the levels were 1.4-fold and 1.2-fold in HSF1-knockdown U87cells, respectively. In addition, HSF1 knockdown significantly suppressed BIS induction upon heat shock. At 6 h of recovery, BIS mRNA expression was 6.6-fold increase in control cells while it was 0.7-fold increase in HSF1-depleted A549 cells. However, effective suppression of BIS did not cause any significant alteration of induction of these two HSP mRNAs in U87 cells, as observed in A549 cells. Thus, BIS was the downstream target for HSF1 transcription factor, but BIS had no ability to cross-regulate HSF1 activity especially in a knockdown model, irrespective of cell type.
Promoter activity of HSP70 and BIS was not dependent on BIS expression levels
We next measured the promoter activity of BIS and HSP70 using a luciferase reporter assay in BIS WT and BIS-KO-A549 cells. The aim of the reporter assay was to ensure that the promoter activity mediated through HSF1-HSE interaction was not influenced by BIS depletion, and to further determine if BIS specifically regulated its own promoter without affecting other HSF1-target gene via an HSF1-independent mechanism. Fig. 3A shows the relative locations of HSEs in the 5′-UTRs of the BIS gene and HSP70 gene. The basal luciferase activity using the HSP70 promoter containing two putative HSEs in WT A549 cells was comparable to that of the KO-A549 cells (Fig. 3B). Furthermore, even after exposure to heat shock stress, the luciferase activity driven by the HSP70 promoter was not reduced in BIS-KO A549 cells compared to WT A549 cells. Similarly, the activity of the BIS promoter with three HSEs was not significantly different between the WT A549 cells and BIS-KO A549 cells before or after heat shock stress. When a series of reporter constructs of various 5′-UTR lengths of the BIS gene were expressed in control A549 cell or BIS knockdown A549 cells, the luciferase activities from all the constructs tested revealed no significant difference between control and BIS-KD A549 cells, regardless of the presence of any putative HSE or length of the BIS promoter region (Figs. 3C and D). Therefore, the reporter assay results suggested that the promoter activity driven by the HSF1-HSE axis was not influenced by BIS depletion, correlating with the expression levels of target genes as shown in Figs. 1 and 2. These results also suggest that alteration of promoter activity did not contribute to the autoregulation circuit of BIS expression at the constitutive or inducible level.
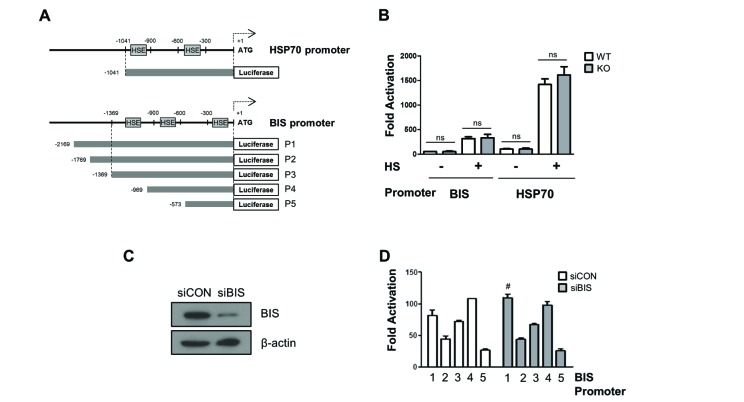
Fig. 3
The promoter activities of BIS and HSP70 were not affected by BIS depletion.
(A) Schematic diagram for the relative position of heat shock element (HSE) to ATG in the promoter region of the HSP70 and BIS genes. (B) WT A549 and BIS-KO A549 cells were transfected with BIS or HSP70 promoter constructs for 24 h and then exposed to heat shock (HS) at 43℃ for 30 min, and then further at 37℃ for 1 h. Transcriptional activation of the BIS (P3) and HSP70 promoters are presented as fold changes compared to the value of the pGL3 basic construct in untreated conditions in WT and BIS-KO A549 cells. (C) Western blotting shows that BIS protein expression was sufficiently repressed by siRNA. (D) Following suppression of BIS in A549 cells with BIS siRNA (siBIS), the reporter constructs were expressed and compared to the control (siCON). After normalization with renilla activity, the results are presented as fold change compared to the activity from the pGL3 basic vector (mean±SE, n=3). #p≤0.05 vs. siCON for P1. ns, statistically non-significant.
![]()
Autoregulation of BIS mRNA expression was not attributable to an altered regulation of mRNA stability
It has been shown that an aberrant mRNA with a truncated open reading frame and a long 3′-UTR due to the presence of a premature stop codon was recognized and degraded by specific mRNA degradation systems, called nonsense-mediated decay. The nonsense-medicated decay pathway is a known translation-coupled mRNA quality control system that prevents cells from producing potentially deleterious truncated proteins, by recruiting a set of factors that destabilize a target RNA [3536]. Therefore, it was possible that the decreases of BIS mRNA in BIS-KO A549 cells were the consequence of nonsense-mediated BIS mRNA decay due to the premature stop codon (Fig. 4A), and were not related with the absence of BIS protein. To confirm this hypothesis, we measured the BIS mRNA stability by an actinomycin D chase assay in WT and BIS-KO A549 cells. As determined by the level of remaining BIS mRNA, determined using qRT-PCR, at the indicated times following treatment with actinomycin D, the degradation rate of BIS mRNA was not notably different between WT A549 cells and BIS-KO A549 cells; t1/2=2.54 h and 2.44 h in WT and BIS-KO A549 cells, respectively. From these results, we found that the regulation of BIS mRNA stability, regardless of 14-bp deletion, was not an essential mechanism which conferred autoregulation of BIS expression. In addition, when the WT BIS gene and deletion mutant of the BIS gene (Δ14-BIS) were expressed as a GFP-fusion construct in WT-A549 cells, the GFP mRNA expression levels were not notably different between the two cell types transfected with each construct (Fig. 4C). Taken together, the deletion of 14 nucleotides in the BIS gene was not responsible for the decrease in BIS mRNA levels in BIS-KO cells. Finally, we tested the BIS pre-mRNA levels in WT A549 and BIS-KO A549 cells using specific primers for intron 2. As shown in Fig. 4D, the BIS pre-mRNA levels represented by intron 2 levels were higher in BIS-KO A549 cells than those in WT A549 cells, while BIS mature mRNA levels determined by the primers covering exon2 and 3 were decreased in BIS-KO A549 cells as measured by primers for exon4 (Figs. 1, 2, 3). Thus, these findings suggest the possibility that splicing process of BIS pre-mRNA may be retarded by BIS depletion.

Fig. 4
Autoregulation of BIS was not mediated by the alteration of mRNA stability.
(A) The sequencing results of genotyping in BIS-KO A549 cells show the deletion in exon 1 of the BIS gene and consequent generation of a premature stop codon. (B) WT and BIS-KO A549 cells were treated with actinomycin D for up to 24 h. At the indicated times, mRNA was extracted, and the remaining BIS mRNA was measured and compared to the initial value with qRT-PCR analysis (mean±SE, n=3). (C) The coding region of the WT BIS and the deletion mutant of the BIS gene, in which 14 bp were deleted (Δ14), were cloned into the pEGFP-C1 construct (left). After transfection for 48 h, the GFP mRNA levels representing WT and Δ14-BIS were determined with PCR amplification and agarose electrophoresis by loading different quantities of PCR products (right). (D) The BIS pre-mRNA levels were determined in WT A549 and BIS-KO A549 cells using qRT-PCR analysis with specific primers for intron 2 (solid arrows). The BIS mature mRNA was levels were also determined by the primers covering exon 2 and 3 (dotted arrows). Reverse transcription was performed as described in Method section. *p≤0.05 vs. WT.
![]()
DISCUSSION
The expression of BIS, a pro-survival protein, is mainly regulated at the transcriptional level by HSF1 in response to various forms of stress [22232425]. Recent studies have shown that BIS physically binds to HSF1 [28], but the effect of the interaction of these two proteins on each other's function has not been clearly defined. BIS was shown to exert diverse effects on cell fates through interaction with various proteins, which affect their stability, localization and activity [3]. Therefore, it is probable that BIS might modulate either the translocation or the transcriptional activity of HSF1. Supporting this possibility, the effect of BIS on nuclear translocation of HSF1 has been previously reported. For example, oxidative stress-induced phosphorylation of BIS protein weakens its binding ability to HSF1, thereby accelerating its nuclear translocation in A172 cells [37]. BIS was also shown to co-translocate into the nucleus with HSF1 upon heat shock stress in HeLa cells [38]. In addition, under glioblastoma stem cell like sphere-forming conditions, BIS depletion is known to decrease HSF1 protein levels and its nuclear localization [39]. Collectively, these results indicate that BIS might be positively involved in the nuclear translocation of HSF1. In contrast, the effects of BIS depletion on the transcriptional activity of HSF1 are not consistent. In the present study, we clearly demonstrated that BIS depletion did not affect the activation of HSF1 as evidenced by the expressions of HSP70 and HSP27 mRNA as well as HSP70 promoter activity in A549 lung cancer cells. However, a previous report showed that adenovirus-mediated suppression of BIS decreased HSP70 and HSP27 protein expression in T98G glioblastoma cells [27]. On the other hands, HSP70 expression was significantly increased in BIS-KO cardiomyocytes [40]. The molecular basis for this inconsistency between our findings and those of previous reports are uncertain. The viral transfection method can create a cell environment in which the host cell is more dependent on the BIS in activating HSF1. It is also likely that BIS depletion either caused or aggravated the accumulation of protein aggregates, thereby secondarily activating HSF1 in cells vulnerable to proteotoxic stress. These results show that nuclear translocation of HSF1 may be affected by the interaction with BIS, which is not necessarily linked to alteration of transcriptional activity of HSF1 on the stress-inducible target genes.
A possible explanation for the lack of functional association of BIS with HSF1 activity shown in our study could have resulted from the application of limited forms of stress such as proteotoxic stress and canonical target genes including HSPs. In addition to proteotoxic stress, HSF1 and subsequent heat shock response are known to be influenced by the proliferative and metabolic state of the cell, which can provide protein homeostasis according to cellular demands [4142]. Thus, BIS might be involved in the activation of HSF1 under specific conditions other than proteotoxic stress. Furthermore, even in unstressed conditions, HSF1 plays an important role in the diverse biological processes including development, metabolism and carcinogenesis through a transcriptional program that is uncoupled from heat shock response [4344]. Therefore, it is also possible that BIS might participate in an alternative transcriptional program that leads to long-term cell survival, which is supported by the constitutive high expression of HSF1 and BIS in various type of human cancer [344445]. However, to investigate the mechanistic network between BIS and HSF1 in the pro-growth program in cancer cells, further studies should be performed under more detailed and diverse experimental conditions.
In the present study, we also demonstrated that the induction profile of the BIS gene, which includes HSEs in the promoter, did not parallel that of the other HSF1-target genes in BIS-KO A549 cells. Considering that BIS expression was primarily regulated by HSF1 upon stress or ectopic expression of BIS itself, our findings were unexpected. Thus, autoregulation of BIS expression may involve separate mechanisms; an HSF1-dependent positive feedback pathway triggered by BIS overexpression and HSF1-independent suppression of its own transcript mediated by BIS depletion. Our findings show that neither promoter activity nor mRNA stability of BIS was affected by BIS depletion. Therefore, the nuclear or cellular events linking these two processes, including splicing process, should be further studied to investigate the molecular mechanism by which BIS controls the autoregulatory loop. Targeting the auto-regulatory pathway is an efficient strategy to suppress BIS expression, which is aberrantly expressed in most cancers.
 XML Download
XML Download