

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pulmonary hypertension (PH) is a progressive and fatal disease characterized by a sustained elevation of pulmonary arterial pressure and pathological changes involving endothelial injury, medial hypertrophy, adventitial thickening, inflammatory cell infiltration, and fibrosis [1]. These remodeled and obstructed vessels persistently provide some limitation of blood flow through pulmonary arteries, which cause an increased right ventricular (RV) afterload followed by RV hypertrophy and failure [1]. Pulmonary vascular remodeling has become a key target for therapy. Although current therapies improve symptoms and hemodynamics of PH, true reversal of pulmonary vascular remodeling is rarely achieved. Therefore, further studies are needed to develop more effective therapies to target the abnormal pulmonary vasculature.
The renin-angiotensin system (RAS) plays an important role in lung pathophysiology because all major components of RAS are expressed in the lung tissue [2]. Particularly, the counter-regulatory arm of RAS, angiotensin converting enzyme (ACE)-angiotensin II (Ang II)-angiotensin type 1 receptor (AT1R) axis and ACE2-Ang-(1-7)-Mas receptor axis, has been highlighted in lung injury. Ang-(1-9) generated from Ang I by ACE2 is known as another counter-balancing axis of RAS. However, little is known about the biological effects of Ang-(1-9) in PH. Available evidence shows that Ang-(1-9) has beneficial effects in preventing/ameliorating cardiovascular remodeling [3]. It has been demonstrated that Ang-(1-9) attenuates cardiac fibrosis through decreasing collagen I expression in stroke-prone spontaneously hypertensive rats [3] and reduces blood pressure with an improvement of endothelial function in renal hypertensive rats [4]. Blockade of the Rho-associated protein kinase (ROCK) signaling pathway reduces blood pressure and vascular remodeling with increasing plasma Ang-(1-9) level and aortic ACE2 activity [5]. Ang-(1-9) also augments nitric oxide (NO) and arachidonic acid releases in endothelial cells leading to enhance the effects of bradykinin and finally improve vascular functions [6]. In addition, the AT2R is known to oppose many of the detrimental actions of the AT1R. Bruce et al. reported that a recently identified non-peptide AT2R agonist, Compound 21 attenuates the progression of PH and inhibits cardiopulmonary fibrosis [7]. Until recently, the effects of AT2R activation have been difficult to ascertain due to lack of effective agonists that can selectively activate AT2R rather than AT1R. The biological effects of Ang-(1-9) are known to be mediated via AT2R [489]. Taken together, we hypothesized that Ang-(1-9) has beneficial effects in PH. The aim of the present study was to investigate the protective role of Ang-(1-9) on vascular remodeling, and its signaling pathway in monocrotaline (MCT)-induced PH rats.
Go to : 
METHODS
Animals
Male Sprague-Dawley rats weighing 200–220 g were purchased from Daehanbiolink Co. Ltd. (Eumsung, Korea) and housed in a temperature-controlled room with a 12:12-h light-dark cycle. Animals were provided free access to standard laboratory chow (5L79 Purina rat & mouse 18% chow, Charles River Laboratories Inc., Wilmington, MA) and water. All experimental protocols conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication No. 85-23, revised 1996) and were approved by our institution.
Experimental protocols
Experiment was conducted with five groups (n=8 for each group).
Group 1 was control rats received saline. Group 2 was MCT-induced PH rats. Adult male Sprague-Dawley rats weighing 200–220 g received a single subcutaneous injection of 50 mg/kg MCT (Sigma-Aldrich Co., St Louis, MO) to induce PH. Group 3 was MCT-induced PH rats pretreated with Ang-(1-9) (Bachem, Switzerland). Ang-(1-9) was infused 3 days before MCT injection for 24 days at a dose of 576 µg/kg/day via a mini-osmotic pumps (Alzet 2004, Cupertino, CA) implanted subcutaneously between the scapulars [10]. Three days after the start of Ang-(1-9) infusion, MCT was injected. Groups 4 and 5 were MCT-induced PH rats pretreated with Ang-(1-9) in the presence of AT2R or Mas R antagonist. Either PD 123,319 (576 µg/kg/day; Sigma-Aldrich Co.) [3] or A779 (576 µg/kg/day; Bachem) [10] was infused via miniosmotic pumps 2 days before the start of Ang-(1-9) infusion, and Ang-(1-9) was then co-administered 2 days later. Three days after the start of Ang-(1-9) infusion, MCT was injected. All rats were sacrificed 21 days after MCT injection.
Measurements of right ventricular systolic pressure and weight
To measure the right ventricular systolic pressure (RVSP) on 21 days after MCT injection, rats were anesthetized with ketamine (Yuhan, Korea) and xylazine (Bayer Korea, Korea). Polyethylene tube connected to PowerLab (AD Instruments Pty. Ltd, Australia) via the pressure transducer (Statham Instruments, Oxnard, CA) was inserted into the RV through the right jugular vein. After stabilization, RVSP was measured for 10 min and blood was collected from the abdominal aorta [11]. Hearts and lungs were quickly removed, weighed, and stored at −70℃ until assayed.
Measurements of hormones and cytokine concentrations in plasma
Plasma atrial natriuretic peptide (ANP) was extracted using a Sep-Pak C18 cartridge and the ANP concentrations in plasma and ventricles were measured using specific radioimmunoassay (RIA), as described previously [12]. The plasma renin concentration (PRC) was also measured using RIA, as described previously [12]. Severity of lung injury was determined by the concentration of lactate dehydrogenase (LDH) using LDH ELISA kit (Takara Bio Inc., Japan) [1314]. Concentrations of tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein-1 (MCP-1), IL-1β and IL-6 in plasma were measured by using rat magnetic bead panel (EMD Millipore, Temecula, CA).
Histologic analysis
Left upper lung tissues were fixed with 4% paraformaldehyde, paraffin-embedded, and sliced with 4 µm thickness. Lung tissue sections were stained with hematoxylin and eosin (H&E). To determine the extent of collagen deposition in pulmonary arteries, Sirius red staining and Masson trichrome staining was performed and quantified by modified ashcroft scoring system [1516]. For immunohistochemisty, the tissue sections were blocked with peroxidase blocking agent for 5 min, washed with Tris-HCl with Tween (TBST), and blocked with protein blocking serum free buffer for 5 min. Then, sections were incubated with anti-α-smooth muscle actin (α-SMA) (Sigma-Aldrich Co.) or anti-von Willebrand Factor (vWF) (Millipore, Temecula, CA) overnight at 4℃ followed by washing with TBST for 5 min. Enough labelled polymer conjugated with secondary antibodies (Dako, Carpinteria, CA) were applied to the slides for 30 min, followed by washing with TBST for 5 min. Peroxidase activity was detected with the ready-to-use AEC+substrate chromogen (Dako). At least twenty arteries of 15-100 µm per each rat were evaluated in α-SMA stained slides through a light microscope (imager M1, Carl Zeiss, Jena, Germany) at ×400 magnification and quantified by image J software.
The medial wall thickness is calculated as follows:
Wall thickness=(total area of artery–lumen area of artery)/total area of artery
Western blot analysis
Lung tissues were homogenized in lysis buffer containing RIPA II cell lysis buffer (GenDEPOT, Barker, TX), protease inhibitors and phosphatase inhibitors and put on the ice for 20 min. These lysed proteins were centrifuged for 15 min. Thirty microgram of protein from supernatant was separated by SDS-PAGE and transferred to PVDF membrane (Bio-Rad Laboratories, Hercules, CA). After blocking with 5% skim milk (Bio-Rad), membrane was incubated with 2 µl primary antibodies against Bax, Bcl-2, Caspase-3, Caspase-9 (Cell Signaling Technology, Denver, MA for all), AT1R (Sigma-Aldrich Co.), AT2R (Sigma-Aldrich Co.), Mas receptor (Alomone Labs, Israel), ACE (Santa Cruz Biochemicals, Santa Cruz, CA), and GAPDH (Bioworld technology, St. Louis Park, MN). After washed with T-TBS buffer 6 times for 5 min each, membrane was incubated with goat anti-rabbit IgG (Enzo Life Sciences) for 30 min. Membrane was then washed again with T-TBS, developed using ECL (Amersham, Arlington, IL) and visualized using LAS-1000 (Fujifilm, Japan).
Statistical analyses
Statistical analyses were performed using GraphPad Prism® 6 (GraphPad Software, La Jolla, CA). The results are presented as the mean±SEM. Statistical significance of the differences was assessed using analysis of variance followed by the Bonferroni multiple comparison test. Student's t test was also used. The critical level of significance was set at p<0.05.
Go to :
RESULTS
Ang-(1-9) ameliorates MCT-induced increases in RVSP and RV weight
The RVSP in MCT-treated rats was significantly higher than that in the normal controls on 21 days after the MCT injection (30.20±0.90 mmHg vs. 13.40±3.30 mmHg; p<0.01) (Fig. 1A). The pretreatment with Ang-(1-9) attenuated the MCT-induced increase in RVSP (19.9±2.8 mmHg), which was inhibited by AT2R antagonist, PD123,319 but not by Mas receptor antagonist, A779 (Fig. 1). Subcutaneous injection of MCT increased the ratio of RV weight to left ventricle+septum (LV+S) weight from 0.26±0.01 to 0.50±0.03 (p<0.01) (Fig. 1B), which was attenuated by the pretreatment with Ang-(1-9) (0.37±0.02). The protective effects of Ang-(1-9) were blunted by the co-treatment of PD123,319 but not by A779 (Fig. 1B). The ANP level, a marker of ventricular hypertrophy, was higher in RV of MCT rats than that in sham rats and the pretreatment with Ang-(1-9) attenuated an increased ANP level in RV (Fig. 1C). PRC did not changed significantly except MCT rats co-treated with Ang-(1-9) and PD123,319 (Fig. 1D). Ang-(1-9) improved the survival rate of MCT-treated rats (83%, n=6 vs. 33%, n=6) on 28 days after MCT injection.
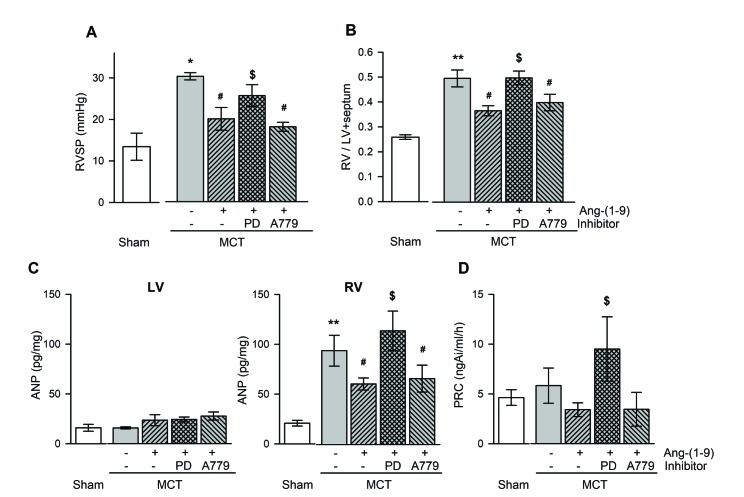 | Fig. 1Effects of Ang-(1-9) on ventricular pressure and weight, and plasma hormone levels in MCT-treated rats.Changes in RVSP (A), RV/LV+septum (B), ANP in ventricular tissue (C), and PRC (D) with and without Ang-(1-9) in MCT-treated rats. RVSP, right ventricular systolic pressure; RV/LV+septum, the ratio of free wall of right ventricular weight to left ventricular weight+septal weight; PRC, plasma renin concentration; MCT, monocrotaline; LV, RV, left and right ventricle, respectively; PD, PD123,319. Values are expressed as mean±SEM of 8 rats per groups. *vs. sham rats, p<0.05, **p<0.01; #vs. MCT rats, p<0.05; $vs. Ang-(1-9), p<0.05.
|
Ang-(1-9) attenuates the endothelial damage and the medial hypertrophy of pulmonary arteriole in MCT rats
Fig. 2A shows a representative immunohistochemical finding of pulmonary arteriole with vWF and α-SMA antibodies, a marker of endothelial damage and medial hypertrophy, respectively. Ang-(1-9) attenuated the expressions of vWF and α-SMA, which were blunted by the co-treatment of PD 123,319, but not by the co-treatment of A779. The quantitative analysis of immunohistochemical finding shows that the medial thickness of pulmonary arterioles was increased in both arterioles sized 50–100 µm and less than 50 µm (external diameter) by MCT treatment and Ang-(1-9) attenuated the medial hypertrophy. The anti-hypertrophic and anti-proliferative effects of Ang-(1-9) on pulmonary arterioles were inhibited by the co-treatment of PD 123,319, but not by the co-treatment of A779 (Fig. 2B).
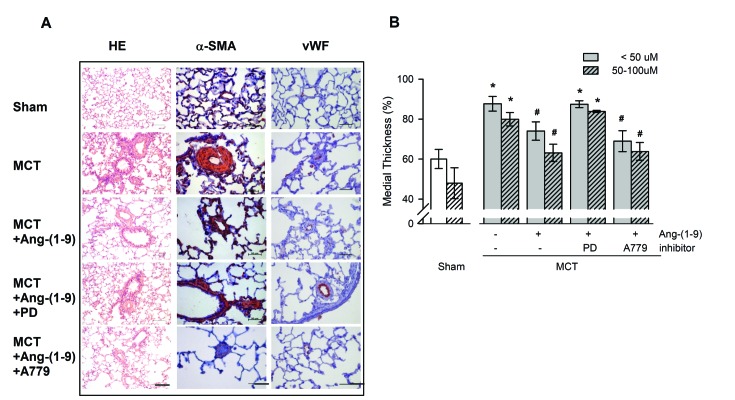 | Fig. 2Effects of Ang-(1-9) on MCT-induced pulmonary arterial intimal proliferation.(A) Representative H&E staining (bar=100 µM) and immunohistochemical staining for α-SMA and vWF (bar=50 µM) in lung tissues of MCT rats with or without Ang-(1-9). (B) Quantification of the medial thickness of pulmonary vessels <50 µm and 50–100 µm in external diameter. Values are expressed as mean±SEM of approximately 30 arterioles of 5 rats per groups. α-SMA, α-smooth muscle actin; vWF, von Willebrand Factor. *vs. sham rats, p<0.05; #vs. MCT rats, p<0.05.
|
Ang-(1-9) reduces inflammation-related cytokines in plasma of MCT rats
Subcutaneous single injection of MCT caused severe pulmonary inflammation. As shown in H&E staining (Fig. 2A), inflammatory cells were dramatically infiltrated in lung of MCT-treated rats compared with that of sham rats. Therefore, we measured ANP and LDH as an indicator of lung injury and disease activity [1314] and pro-inflammatory cytokines [17] in plasma (Fig. 3). The plasma concentration of ANP tended to increase without significance in MCT rats and was higher in Ang-(1-9)-pretreated group (Fig. 3A). The levels of LDH, MCP-1, IL-1β, IL-6, and TNF-α were significantly increased in plasma of MCT rats compared with those of sham rats. These increases were attenuated by Ang-(1-9) administration. The anti-inflammatory effect of Ang-(1-9) tended to be blocked by the co-treatment of PD123,319.
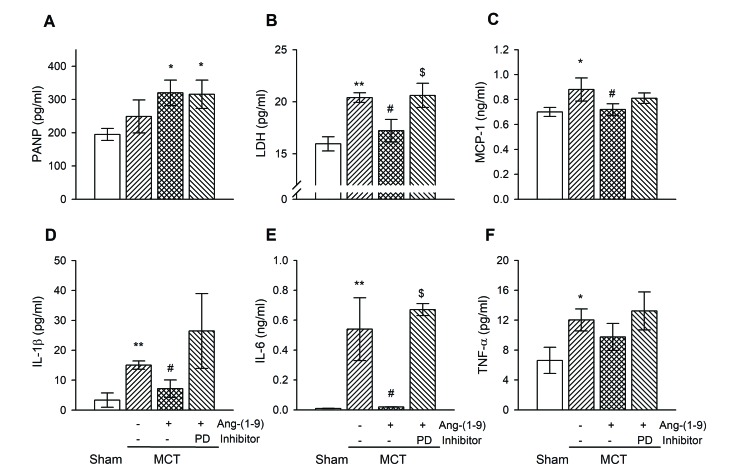
Ang-(1-9) attenuates lung fibrosis in MCT rats
To determine the extent of collagen deposition in pulmonary arteries, Sirius red staining and Masson trichrome staining were performed and quantified by modified ashcroft scoring system [15]. Most of the lung structures were damaged and more collagen was deposited compared with those of sham (Fig. 4A). Not only in perivascular region but in pulmonary interstitium, alveolar septal walls were severely thickened and confluent fibrotic masses were seen in MCT-treated rats. Ang-(1-9) administration decreased adventitial fibrosis and collagen deposition without fibrotic masses. Lung sections of PD123,319 with Ang-(1-9) administration group were shown similar characteristics as those of MCT rats, demonstrating the protective effect of Ang-(1-9) is via AT2R. By quantitative analysis of immunohistochemistry, MCT rats showed high grade of lung fibrosis which was inhibited by Ang-(1-9) treatment (Fig. 4B). The anti-fibrotic effect of Ang-(1-9) was abolished by the co-treatment with PD123,319 but not by the co-treatment with A779.
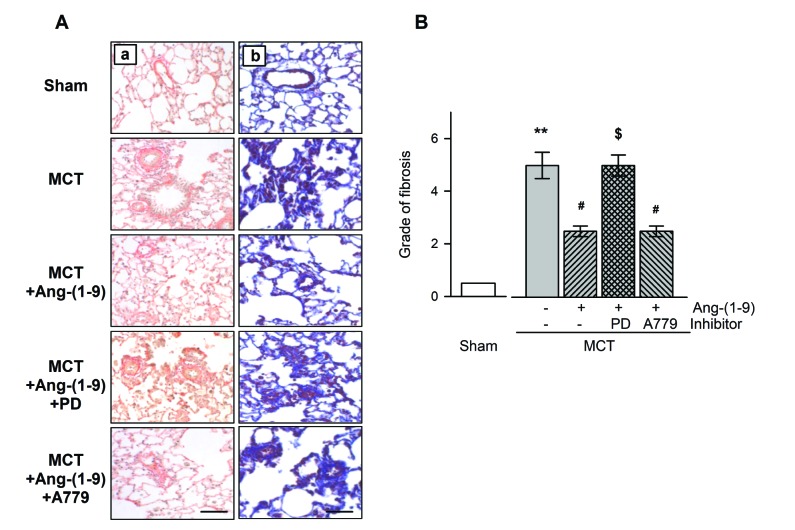 | Fig. 4Effects of Ang-(1-9) on MCT-induced pulmonary fibrosis.Immunohistological analysis of pulmonary fibrosis using Sirius-red (Aa), Trichrome masson (Ab) staining (bar=100 µM) and quantification by modified ashcroft scoring system (B) with or without Ang-(1-9). Values are expressed as mean±SEM of 8 rats per groups. **vs. sham rats, p<0.01; #vs. MCT rats, p<0.05; $vs. Ang-(1-9), p<0.05.
|
Ang-(1-9) changes the expression of apoptotic-related proteins and Ang-related receptors in lung tissues of MCT rats
To evaluate whether the protective effect of Ang-(1-9) in MCT rats is mediated via apoptosis, the expressions of apoptotic-related proteins in lung tissues were measured. Western blot showed that the expressions of Bax, caspase-3, and -9 proteins were increased and the expression of Bcl protein was decreased in lung tissues of MCT rats (Fig. 5A). The changes in apoptotic-related protein expressions in MCT-treated rats were attenuated by the pretreatment of Ang-(1-9) (Fig. 5B). The anti-apoptotic effect of Ang-(1-9) tended to be attenuated by the co-treatment with PD123,319.
 | Fig. 5Effects of Ang-(1-9) on apoptotic-related protein expressions in lung tissues of MCT rats.Representative western blotting of Bax, Bcl-2, Caspase-3, and Caspase-9 protein expressions in lung tissues of MCT rats with or without Ang-(1-9) (A) and quantification of protein expressions compared to GAPDH (B). Values are expressed as mean±SEM of 8 rats per groups. V, vehicle; PD, PD123,319; A, A779. *vs. sham rats, p<0.05, **p<0.01; #vs. MCT rats, p<0.05.
|
MCT injection increased AT1R and Mas R expressions and decreased AT2R expression in lung tissue, which was reversed by the pretreatment with Ang-(1-9) infusion (Fig. 6). These effect of Ang-(1-9) tended to be attenuated by the co-treatment with PD123,319. However, a decreased expression of ACE in MCT rats tended to increase without significance by the pretreatment with Ang-(1-9).
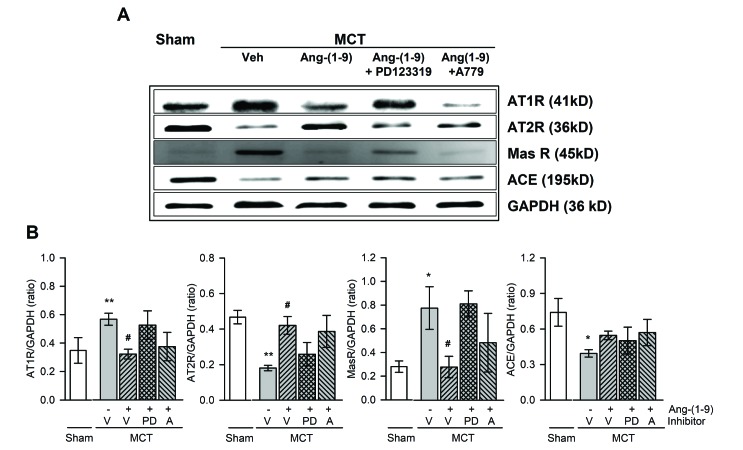 | Fig. 6Effects of Ang-(1-9) on angiotensin-related protein expressions in lung tissues of MCT rats.Representative western blotting of angiotensin type 1 receptor (AT1R), AT2R, Mas receptor (Mas R), and angiotensin converting enzyme (ACE) protein expressions in lung tissues of MCT rats with or without Ang-(1-9) (A) and quantification of protein expressions compared to GAPDH (B). Values are expressed as mean±SEM of 8 rats per groups. V, vehicle; PD, PD123,319; A, A779. *vs. sham rats, p<0.05, **p<0.01; #vs. MCT rats, p<0.05.
|
Go to :
DISCUSSION
We found that Ang-(1-9) attenuated the progression of PH. The administration of Ang-(1-9) ameliorated not only RV hypertrophy but pulmonary vascular remodeling induced by MCT treatment. Increases in inflammatory cytokines in plasma and in apoptotic-related protein expressions in the lung were also attenuated by Ang-(1-9). These beneficial effects of Ang-(1-9) were blunted by co-treatment with AT2R antagonist. Therefore, we suggest that the protective effects of Ang-(1-9) against PH are mediated via AT2R through an inhibition of apoptosis and inflammation.
Ang-(1-9) can be detected in plasma and its circulating level increased early after myocardial infarction or in animals treated with ACE inhibitor [18]. Available evidence shows that Ang-(1-9) has beneficial effects in preventing/ameliorating cardiovascular remodeling [419]. However, the effects of Ang-(1-9) on PH are not clear. Experimental PH models induced by chronic hypoxic exposure and MCT treatment have been used to investigate the pathogenesis and treatment of human PH. MCT-induced PH model has some advantage for the understanding of vascular remodeling [20] compared with chronic hypoxia-induced PH model [21]. The most common features of PH are vascular remodeling characterized by endothelial injury and medial wall thickening, and perivascular inflammation and fibrosis. Especially, wall thickening of pulmonary arterioles leads to increase a pulmonary vessel resistance and RV afterload followed by RV hypertrophy. In the present study, MCT administration resulted in a significant increase in RVSP, which was associated with the development of RV hypertrophy. The immunohistochemical finding of pulmonary arteriole stained with vWF and α-SMA antibodies showed severe endothelial damage and medial hypertrophy of pulmonary in MCT-treated rats. The pretreatment with Ang-(1-9) reduced the expression of vWF and α-SMA. In lining with this result, both RVSP and RV hypertrophy were attenuated by Ang-(1-9). The pretreatment with Ang-(1-9) attenuated an increased ANP level, a marker of ventricular hypertrophy, in RV. Ang-(1-9) is reported to mediate its biological effects via AT2R [822]. The beneficial effects of Ang-(1-9) were blunted by the co-treatment with AT2R antagonist but not by Mas R antagonist. These results suggest that Ang-(1-9) ameliorates PH via AT2R. Our data are partly consistent with other reports showing beneficial effects in ameliorating cardiovascular remodeling [4519].
Ocaranza et al. have reported that Ang-(1-9) reduces hypertension, ameliorates structural alterations, and improves cardiac and endothelial function in hypertensive rats [4]. Inhibition of Rho kinase pathway increases aortic ACE2 activity and Ang-(1-9) plasma levels in DOCA hypertensive rats leading to reducing blood pressure and vascular remodeling [5]. The same group also reported that Ang-(1-9) attenuates cardiac hypertrophy via AT2R in myocardial infarcted rats [19]. Flores-Munoz et al. suggest that Ang-(1-9) antagonizes pro-hypertrophic signalling in cardiomyocytes [9] and attenuates cardiac fibrosis in the stroke-prone spontaneously hypertensive rats [3] via the AT2R, proposing Ang-(1-9) is an independent counter-balanced arm of the RAS. Most of studies with Ang-(1-9) described the above have been done using spontaneously, renal, DOCA- or Ang II-induced hypertensive rats to define the cardio-protective effects. The protective effects of Ang-(1-9) against PH are comparable with report showing attenuation of PH and inhibition of cardiopulmonary fibrosis by AT2R agonist [7].
Recent studies suggest that inflammation contributes to the development of PH and the degree of perivascular inflammation is related to both vascular remodeling and increasing mean pulmonary arterial pressure [23]. Inflammation consists of both the presence and the activity of professional inflammatory cells via growth factors and cytokines. T cells and macrophages are mainly found in plexiform lesions of lung tissue in PH patients [24] and mRNA levels of pro-inflammatory cytokines are elevated in the lung of MCT-treated rats [25]. The results showing an increased serum levels of TNF-α and IL-6 in both MCT-treated rats and PH patients suggest that elevated levels of inflammatory cytokines may predict a survival rate in PH [26]. In addition, IL-6 accelerates the progress of PH [27], and the TNFα antagonist prevents and reverses MCT-induced PH [28]. Our studies showed an infiltration of inflammatory cells, especially macrophages and monocytes, in the lung and high levels of pro-inflammatory cytokines, such as MCP-1, TNF-α, IL-1β, and IL-6 in plasma of MCT rats. The pretreatment with Ang-(1-9) inhibited the infiltration of inflammatory cells and decreased plasma levels of cytokines and LDH. These observed results suggest that the protective effect of Ang-(1-9) is partly related to inhibition of inflammation. Ang II is known to be involved in perivascular inflammation through the generation of ROS and the release of pro-inflammatory cytokines [29] whereas overexpression of Ang-(1-7) prevents an increase of pro-inflammatory cytokines [25]. The beneficial effects seen with ACE2/Ang-(1-7)/Mas receptor activation are similar to those seen by AT2R activation with Compound 21 in MCT rats [7]. However, in the present study, we did not know the localization of AT1R and AT2R. I has been reported that AT1R and AT2R is localized in the endothelium and smooth muscle cells of pulmonary artery. Our data are consistent with the report showing that in PH, the expression of AT1R but not AT2R is upregulated [3031]. Taken together the above results, the ACE2/Ang-(1-9)/AT2R axis similarly to ACE2/Ang-(1-7)/Mas receptor is another counter-regulatory arm of RAS to ACE/Ang II/AT1R.
Apoptosis from lung parenchymal damage and pulmonary endothelial cells [32] has been well recognized in the MCT-induced PH in recent studies [33]. In this study, apoptosis-related protein levels were measured by western blot. Consistent with the findings of previous reports [3334], we demonstrate that the expressions of apoptotic biomarkers were substantially increased in lungs of MCT-treated animals and Ang-(1-9) attenuated the highly expression of apoptotic proteins. Therefore, we suggest that the anti-apoptotic effects of Ang-(1-9) might partly relate to an inhibition of the development of PH.
In conclusion, Ang-(1-9) reduced RVSP and RV hypertrophy in MCT-induced PH. Ang-(1-9) inhibited the infiltration of inflammatory cells and reduced the pro-inflammatory cytokines. These beneficial effects of Ang-(1-9) were mediated by AT2R. Taken together, Ang-(1-9) may act as a counter-balancer of Ang II, so can be a therapeutic candidate of PH.
Go to :
 XML Download
XML Download