

 PDF
PDF ePub
ePub Citation
Citation Print
Print


서론
유전구형적혈구증(hereditary spherocytosis)은 적혈구의 spectrin 단백과 이를 적혈구 막과 부착시키는 단백인 ankyrin, band 3, protein 4.2의 결함에 의해 적혈구가 구형을 띠게 되고, 삼투압 취약성이 증가하며, 역학적 안정성(mechanical stability)이 떨어져 비장에서 파괴되고 용혈을 일으키는 가장 흔한 유전용혈성빈혈이다. SPTA1, SPTB, ANK1, SLC4A1, EPB42 유전자의 돌연변이에 의한 α spectrin, β spectrin, ankyrin, band 3, protein 4.2의 양적, 질적 6338이상에 의한 유전구형적혈구증이 보고되어 있으며[1234], 약 75%에서 상염색체 우성으로 유전되나, 나머지에서는 우성유전방식을 따르지 않으며(nondominant), 상염색체 열성 또는 de novo로 발생한 경우가 보고되어 있다[5].
Band 3는 주요한 적혈구 막의 내재성 단백으로 anion exchanger 1 (AE1)으로도 불린다[6]. Band 3는 chloride/bicarbonate exchanger인 membrane-spanning domain, C-terminal, N-terminal cytoplasmic domain의 3개의 domain으로 이루어져 있으며, N-terminal domain의 주요기능은 적혈구 막을 세포골격에 고정시키는 것이다. 적혈구 막에서 band 3는 이합체 및 사합체로 존재하는데, 사합체가 ankyrin과 protein 4.2와 결합하게 되면서, 적혈구 막이 세포골격에 부착하게 된다. 구형적혈구증 4형(Spherocytosis, type 4, OMIM 612653)은 band 3 단백의 해당 유전자인 SLC4A1의 돌연변이에 의해 발생한 유전구형적혈구증을 말하며, 구형적혈구증 환자의 15–20%에서 발견된다고 알려져 있으나[7], 국내 연구에서는 5% 정도로 낮다[8]. 최근 저자들은 구형적혈구증 환자에서 SLC4A1 유전자 돌연변이 검출을 통해 유전구형적혈구증을 확진할 수 있었던 2예를 보고하고자 한다.
증례
1. 환자 1
특별한 가족력, 과거력이 없는 3세 여아가 3일간 지속된 발열과 상기도감염 증상을 주소로 내원하였다. 내원 당시 시행한 일반혈액검사 결과, 혈색소 10.0 g/dL, 적혈구용적률 27.3%, 백혈구 19.64 ×109/L, 혈소판 216×109/L였으며, 평균적혈구용적 82.5 fL, 평균적혈구혈색소 30.2 pg, 평균적혈구혈색소농도 36.6%, 적혈구크기분포폭 17.3%, 망상적혈구 9.14% (절대 망상적혈구 수 320×109/L)이었다. 말초혈액도말검사에서 적혈구부동증 및 구형적혈구가 관찰되었다. 직간접 쿰스검사 모두 음성소견을 보였으며, 삼투압취약성 검사상 용혈 시작 농도는 0.55% NaCl, 완전 용혈 농도는 0.35% NaCl이었으며, 대조군에서는 0.45%, 0.25%로 대조군보다 약간 증가한 소견을 보였다. 24시간 항온 후 시행한 검사에서는, 용혈 시작 농도는 0.80%, 완전 용혈 농도는 0.40%이었으며, 대조군에서는 0.55%, 0.30%로 삼투압취약성이 대조군보다 증가했음을 확인할 수 있었다. 총 빌리루빈은 2.25 mg/dL, 직접(포합) 빌리루빈은 0.67 mg/dL이었으며, 젖산탈수소효소가 739 U/L로 증가한 소견을 보였다. 복부 초음파 결과 경한 비장종대 소견을 보였으며, 비장크기는 9.5 cm로 2–4세 소아 비장크기의 90th percentile (8.6 cm) [9] 이상이었다. 적혈구 막단백 검사는 시행하지 않았으며, 말초혈액도말의 구형적혈구 및 삼투압 취약성의 증가, 간접빌리루빈 증가, 젖산탈수소효소의 증가와 비장종대 소견을 고려해 볼 때 유전구형적혈구증에 합당한 소견이었다.
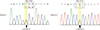
국내에서 빈도가 높은 유전구형적혈구증 관련 유전자인 ANK1, SPTB 유전자를 우선적으로 직접염기서열검사법(Sanger sequencing)으로 검사하였고, 질병연관변이는 관찰되지 않았다. SLC4A1 유전자의 직접염기서열검사 결과, Exon 17에서 c.2278C>T (p.Arg 760Trp)가 관찰되었다(Fig. 1A). 상기 돌연변이는 이전에 유전용혈성빈혈 환자에서 보고되었던 변이로[101112] (PS1), 웹 기반의 in-silico analanalysis에서 class C65 (Align GVGD, http://agvgd.iarc.fr/), score 1.000 (Polyphen-2, http://genetics.bwh.harvard.edu/pph2/), score 0.00 (SIFT, http://sift.jcvi.org/www/SIFT_enst_submit.html), disease causing (MutationTaster, http://www.mutationtaster.org/)으로 질병연관변이일 가능성이 높을 것으로 예측되었으며(PP3), ExAC (http: //exac.broadinstitute.org)에서 allele frequency가 0.000008325, 1000 Genomes Project (http://1000genomes.org), Korean Reference Genome Database (KRGDB, http://152.99.75.168/KRGDB/menuPages/intro.jsp)에서는 보고된 적이 없었다(PM2). 질병연관변이로 보고된 SLC4A1 유전자의 변이는 과오돌연변이가 대부분을 차지하고(PP2), 상기 변이가 단백질 발현에 영향을 미친다는 기능 연구(functional study)가 보고되어 있다[10] (PS3). 환자의 부모 모두 증상이 없었고, 어머니에서 검사를 시행한 결과 동일한 돌연변이는 관찰되지 않았으며, 아버지는 검사를 시행하지 못했지만, 환자의 변이는 de novo로 예측되었다(PM6). The American College of Medical Genetics and Genomics (ACMG) standards and guidelines for the interpretation of sequence variants에 따르면 상기 변이는 pathogenic으로 예측되었다[13].
기관지폐렴으로 진단하여, 항생제 치료 후 증상이 호전되어 퇴원하였다. 이후, 추적관찰 중인 진단 1년 후, 어지럼증과 간헐적인 발열로 응급실에 내원하였으며, 당시 혈액검사에서 혈색소치 5.3 g/dL로 심한 빈혈 소견을 보였다. 젖산탈수소효소가 증가하였으며(1,301 U/L), parvovirus B19 중합효소연쇄반응과 IgM 항체검사에서 양성 소견을 보여, parvovirus B19에 의해 발생된 골수무형성위기(aplastic crisis)로 진단되었다. 이에 농축적혈구제제 수혈 후, 임상증상이 호전되어 퇴원하였고, 혈색소치는 수혈 없이, 9.0–13.0 g/dL 사이로 유지되고, 망상적혈구는 10% 미만으로 유지되어, 유전구형적혈구증의 중증도 분류[1] 중 mild 혹은 moderate에 속하여 folate 1 mg/day를 복용하며 추적관찰 중에 있다. 최근 혈색소는 12.0 g/dL, 망상적혈구는 5.6%이었다.
2. 환자 2
23세 남자 환자가 2016년 8월 반복되는 복통을 주소로 내원하였다. 환자는 7년 전부터 간헐적으로 황달이 나타났으며, 아버지와 친할아버지는 빈혈이 있으나 유전구형적혈구증에 대한 검사는 시행하지 않았다. 환자의 고모는 유전구형적혈구증으로 비장절제술과 담낭제거술을 받은 과거력이 있었으나 유전자 분석은 시행하지 않았다. 고모의 딸은 환자와 함께 내원하였으며, CBC검사, 말초혈액도말검사 및 삼투압취약성 검사에서 모두 정상소견을 보였다. 내원 당시 시행한 CBC 결과, 혈색소 14.4 g/dL, 적혈구용적률 38.9%, 백혈구 6.15×109/L, 혈소판 162×109/L이었으며, 평균적혈구용적 86.4 fL, 평균적혈구혈색소 32.0 pg, 평균적혈구혈색소농도 37.0%, 적혈구크기분포폭 15.1%, 망상적혈구 3.49% (절대 망상적혈구 수 157×109/L)이었다. 말초혈액도말검사에서 적혈구부동증 및 구형적혈구가 관찰되었다. 직접, 간접 쿰스검사 모두 음성소견을 보였다. 총 빌리루빈은 4.58 mg/dL, 직접(포합) 빌리루빈은 0.40 mg/dL이었으며, 젖산탈수소효소가 415 U/L였다. 복부 초음파에서 12 cm의 비장종대 소견이 관찰되었다. 삼투압취약성 검사와 적혈구 막단백 검사는 시행하지 않았으며, 위 소견을 바탕으로 유전구형적혈구증을 의심하고 유전적 진단을 위해 ANK1, SPTB 유전자의 직접염기서열검사를 시행했으며, 질병연관변이는 관찰되지 않았다. SLC4A1 유전자의 Exon 18에서 이형접합 과오돌연변이 c.2423G>T (p.Arg808Leu)가 검출되었다(Fig. 1B). 이 돌연변이는 보고된 적이 없는 변이로 웹 기반의 in-silico analysis에서 class C65 (Align GVGD) score 1.000 (Polyphen-2), score 0.00 (SIFT), disease causing (MutationTaster)으로 예측되었고(PP3), ExAC, 1000 Genomes Project, KRGDB에서 모두 보고되지 않아(PM2) 질병연관변이일 가능성이 높을 것으로 판단되었다. 또한 SLC4A1 유전자의 808번 아미노산의 변화를 유발하는 다른 과오돌연변이(p.Arg808Cys, p.Arg808His)가 질병유발변이로 보고되어 있으며[1014] (PM5), 보고된 SLC4A1 유전자의 돌연변이의 대부분이 과오돌연변이이고, 양성과오변이(benign missense variation)의 비율이 낮아(PP2) ACMG standards and guidelines for the interpretation of sequence variants에 따라 상기 변이는 likely pathogenic으로 예측되었다[13]. 임상적 의의를 보다 명확히 하기 위해서는 추후 부모 및 증상이 있는 고모의 유전자 검사가 필요할 것으로 판단되었다.
고찰
유전구형적혈구증의 가장 흔한 원인 유전자는 ANK1 (~50%), SPTB (~20%)이며, SLC4A1이 15<20%를 차지한다[7]. 이전 보고에 따르면 한국에서 보고된 유전구형적혈구증 환자의 52%에서 ANK1, 48%에서 SPTB 유전자의 돌연변이가 관찰되었으며[2], SLC4A1 유전자 변이 3예가 보고되어 있다[8]. 환자 1은 경한 빈혈을 보였으며, 환자 2는 진단 시 혈색소치는 정상이었지만 간헐적으로 황달 소견을 보인 과거력이 있었다. 환자 1은 추적관찰 동안 수혈 없이 혈색소 10.0–12.0 g/dL로 유지되었으며, 한 번의 parvovirus B19 관련 골수무형성위기로 수혈을 받았다.
유전구형적혈구증은 적혈구 막의 단백질 결함에 의해 적혈구 막의 골격과 지질 이중층의 결합이 끊어지면서 소포(vesiculation)가 형성되어 막 표면이 소실되고, 표면적이 줄어들어 적혈구형태가 구형을 띠게 된다. 적혈구의 표면적 대 용적 비(surface-to-volume ratio)가 감소하여 적혈구 변형성(deformability)이 떨어지며, 이로 인해 구형 적혈구는 비장삭(splenic cord)에 울혈이 일어나게 되고 울혈된 적혈구는 쉽게 용혈을 일으켜 적혈구의 수명이 단축된다. 이로 인해 비장종대가 관찰되며, 용혈빈혈이 발생한다. 용혈에 의해 빌리루빈이 증가하여 황달이 발생하며 증가한 빌리루빈을 간에서 대사하지 못하여 빌리루빈산염 담석이 생성된다. 이러한 증상은 다양한 정도로 나타나며, 정도에 따라 증상 발현시기가 달라지는데 심한 경우, 임신 중의 태아에서부터 증상이 발현될 수 있다. SLC4A1 이상에 의한 유전구형적혈구증은 대부분 경도 또는 중등도의 유전구형적혈구증을 보이며 태아수종은 드물게 나타나며[15], 서로 다른 변이가 각각의 대립유전자에 위치한 복합이형접합체 환자의 경우, 더 심한 임상증상이 나타날 수 있다[616].
유전구형적혈구증과 관련되어 보고된 SLC4A1 유전자 돌연변이는 과오돌연변이(40%), 틀이동돌연변이(30%)가 대부분을 차지하며, 스플라이싱돌연변이(16%), 무의미돌연변이(10%)와 대량중복(1%)이 보고되어 있다. Band 3 단백은 적혈구뿐 아니라 신장의 distal nephron에 존재하는데 신장의 band 3 단백은 N-terminal의 65개의 아미노산이 결여되어 있는 형태이며, 따라서 SLC4A1 돌연변이에 의해 신세관산증이 발생할 수 있다[17]. SLC4A1 돌연변이로 유전구형적혈구증과 신세관산증이 함께 발생한 환자도 보고되어 있다[18]. 하지만 특정 돌연변이와 두 표현형에 대한 상관관계는 밝혀져 있지 않다. 다른 band 3 돌연변이와는 달리 타원적혈구형태를 특징으로 하며, 적혈구가 견고(rigid)하여 말라리아에 저항성을 보이는 Southeast Asian ovalocytosis (SAO)을 일으키는 돌연변이가 보고되어 있는데[19], 상염색체 우성으로 유전되며, 경한 용혈 소견을 보이거나 용혈 소견이 없다. 드물게 가시적혈구증가증(acanthocytosis) [20]이 보고되어 있으며, 혈액형 관련 변이로 보고된 예가 있다[21].
위 환자들과 같이 임상 소견과 기존의 검사법으로 유전구형적혈구증이 의심될 경우 유전자 검사를 활용한다면 확진이 가능하고, 유전자 혹은 변이의 종류에 따라 임상증상과 예후를 예측하여 치료방향을 설정하고 계획하는 맞춤의학(precision medicine)을 구현할 수 있을 것이다. 위 예처럼 band 3 돌연변이에 의한 유전구형적혈구증은 ankyrin, spectrin 돌연변이의 유전구형적혈구증에 비해 임상증상이 경한 것으로 알려져 있으며, 다른 돌연변이에 의한 유전구형적혈구증과 달리 신세관산증이 함께 발생할 수 있다. 이처럼 유전자 검사를 통한 유전구형적혈구증의 진단은 환자의 추적관찰 및 치료에 활용할 수 있는 중요한 정보를 제공할 수 있다. 더불어 현재 검사실에서 차세대염기서열분석(next generation sequencing, NGS)이 통상적인 검사로 활용되면 Sanger sequencing으로 여러 유전자를 순차적으로 분석했던 검사 절차에 비해 시간과 비용을 단축한 gene panels 검사법이 유전구형적혈구증 환자들을 감별진단하는 데에 효율적으로 사용될 것으로 생각된다.
 XML Download
XML Download