

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
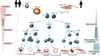
Stem cells (SCs), common to all multicellular organisms, are specified as undifferentiated self-replicating cells possessing the ability to generate, sustain and replace terminally differentiated cells. They show two key features: self-renewal (cell divisions with maintenance of the undifferentiated state), and capability of in vivo and in vitro reconstitution of a given tissue via differentiation into specialized cell types1. SCs are commonly subdivided into two main entities, embryonic stem cells (ESCs) (pluripotent) and adult SCs (multipotent or unipotent) (Fig. 1). A third category of “embryonic-like” cells, so-called induced pluripotent cells (iPSCs), has been added in the last years. iPSCs are developed through genetic manipulation of differentiated cells.
The attributes “pluri-, multi- and unipotent” describe the SC's potential to yield a range of cell lineages. While pluripotent SCs are able to give rise to all cell types in an organism, multipotent and unipotent SCs remain restricted to specific tissue(s) or lineages. The level of potency is linked to the developmental stage of the organism and is evaluated by functional assays and assessment of various cellular/molecular markers1.
Potency defines subsets of stem cells
Totipotent cells are exclusively present in the earliest stadium of embryonic development, mainly during the growth of the fertilized oocyte (zygote) to the eight-cell embryo (i.e., up to five days after fertilization of the egg)23. They possess the capability to generate terminally differentiated cells of the whole trilaminar embryonic disc (ectoderm, mesoderm, and endoderm). Thus, they are able to form an embryo (i.e., a complete human body) as well as extraembryonal tissues such as the placenta along a series of divisions and differentiations. The cells of the early embryo lose their universal potency after the 8-cell stage and a subpopulation of their progeny becomes pluripotent.
Pluripotent stem cells (pSCs) are found in the blastocyt's inner cell mass (ESC) as well as in postnatal adult tissues and are capable of differentiating into all cell types (somatic and germline) of an organism. However, they don't possess the capacity to generate a complete organism. pSCs are defined by their potential to generate embryoid bodies (i.e., non-adherent cell aggregates of pSCs/spheroids) in vitro, and teratomas in vivo. Teratomas are benign tumors comprising endodermal, mesodermal, and ectodermal derivatives, each to a variable extent. Accordingly, embryogenesis and the subsequent phases of development of an organism might be considered as a continual loss of potency2345.
Once SCs are assigned to a specific tissue, they evolve into multi- or unipotent adult SCs, as which they are capable of forming tissues composed of cells at more specialized stages. The variety of differentiated cells that originate from adult SCs is restricted. Adult SCs reside in most organs and tissues (including skeletal muscle, pancreas, heart and central nervous system), but mainly at sites with rapid cell turnover (skin, bone marrow, intestinal mucosa, liver). They reside in (SC) niches with a specific microenvironment necessary for their control and regulation1.
Based on their origin and differentiation capacity, it is possible to categorize adult SCs into many subpopulations. The best investigated are hematopoietic SCs, emanating from the bone marrow. They predominantly generate all hematopoietic tissues and mesenchymal stem/stromal cells, which mainly account for the regeneration of connective tissues, although they are also found in many other tissues (e.g., bone-marrow, adipose tissue, placenta, umbilical cord blood)6.
In 2006, the field of SCs has been augmented by the discovery of iPSCs. They are in vitro manipulated somatic cells, genetically reprogrammed to revert to a state of pluripotency (immature, undifferentiated cells), preceding their current differentiation status. Thereby iPSCs gain the potential to self-renew and subsequently to undergo differentiation into cells of endoderm, mesoderm or ectoderm. This reprogramming process reverts differentiated cells to the undifferentiated stage of ESCs. It is achieved through transfer of various combinations of reprogramming factors, typically including transcription factors like OCT4, SOX2, KLF4, c-MYC (proto-oncogene), NANOG and LIN28, which are known as major factors in the regulation of differentiation and self-renewal of undifferentiated ESCs78910. Up to now, iPSCs have been yielded from several cell types and by various reprogramming methods (retroviral, lentiviral or non-integrating adenoviral vectors11, plasmids12, recombinant proteins13, modified messenger RNAs14, small epigenetic modifier molecules15, transposons16 as well as with different efficiencies. The range of the latter extends in primary reprogramming systems from 0.01% up to 1%, depending on the applied protocol and cell type. This is caused by the different effects of the preexisting gene expression pattern of distinct adult cell types on the forced expression of the reprogramming factors17.
Identification of SCs
Generally, SCs resemble other mature cells and have no unique morphologic feature. They tend to display a high nuclear to cytoplasmic ratio and generate large cell-colonies (a SC with this ability is called “holoclone”) when placed in culture, which reflects their long-term self-renewal and regenerative potential118. Approaches to identify tissue specific SCs include the measurement of quiescence as a typical feature of SCs. This term describes a reversible state of reduced cellular turnover that is actively maintained and regulated by signaling pathways and permits rapid reactivation and reentry in the cell cycle. In addition, most SCs (except for hematopoietic SCs) show a characteristic behavior in culture with formation of tightly compact cell colonies that can be repeatedly passaged and transplanted. In vivo lineage tracing methodology utilizes single SCs permanently marked by e.g. genetic markers, fluorescent proteins, transfection or viral transduction to track labeled (clonic) progenies. Finally, SCs are delineated by the use of specific markers. Their expression profile, however, vary depending on (stem) cell type, their state of activity and anatomical location. Moreover, these markers are rarely unique and need not necessarily be linked to SC function19.
As an example, bone marrow derived mesenchymal stem/stromal cells (BM-MSCs) have no known single, exclusively expressed phenotypic marker. Their isolation from bone marrow or identification in vitro cultures thus relies on a negative selection with depletion of all other BM-cells as well as a combination of SC markers. As for the latter, BM-MSCs are void of hematopoietic and endothelial markers, staining negative for CD45, CD34, all hematopoietic lineage markers, and CD31. Characteristic surface markers of BM-MSCs comprise CD29+, CD73+, CD90+, CD105+, CD106+ and STRO-1+20.
Regulation of stem cell activity
SCs behavior is directed at multiple levels in response to activating, amplifying and inhibiting signals arising from local, environmental and systemic factors. Components of the wingless-type (Wnt)/β-catenin, Sonic hedgehog and Notch, transforming growth factor (TGF-β)/bone morphogenetic protein pathways as well as Nanog, MAPK, c-Myc and p63 receptor kinase cascades are of particular relevance for SC function21.
The Wnt signaling governs major developmental steps in the embryonic state and controls maintenance, self-renewal and differentiation of adult SCs. Wnt pathways are either β-catenin dependent (whereby β-catenin interacts with other transcription pathways, including Sox family members, FOXO, HIFI) or β-catenin independent. Anyway, both cascades have an impact on SC fate in developing and adult tissues22.
Signals downstream of the Wnt pathway can direct a SC in different ways: (i) to stay dormant, (ii) to undergo an asymmetric division (whereby a SC yields two distinct daughter cells, one copy of the original SC and one cell programmed to differentiate), (iii) to exert a non-differentiative symmetric division (generating two copies of the original SC) or (iv) a differentiative division (SC divides into two differentiating cells).
For instance, binding of nuclear β-catenin to the co-activator CREB-binding protein (CBP) forms a transcriptionally active complex that favors symmetric proliferation and preservation of multi(pluri-)potency. In contrast, interaction of β-catenin with nuclear co-activator p300 induces transcriptional sequelae that favor asymmetric SC division and differentiation. These mechanisms are tightly regulated and coordinated by innumberable microenvironmental conditions in the vicinity, including cytokine milieu, vascularity, temperature, niche conditions and occasional presence of toxic materials. SCs are thus continuously receiving a myriad of chemical Wnt-related signals from surrounding cells that themselves again interact with numerous mediators23.
SKIN STEM CELLS
Continuous exposure of the skin to environmental mechanical and chemical stress requires permanent self-renewal of the epidermis, dermis and adnexa (hair follicles [HFs], sebaceous glands, and sweat glands), even into adulthood, to maintain its diverse functions (e.g., as a barrier). This self-renew ability is contributed to the skin own SCs, which are slow cycling multipotent cells located in the epidermis, dermis and the HFs. In response to external stimuli like wounding, they start to proliferate in order to regenerate the skin tissue24.
When primary cultures of keratinocytes are grown in vitro, three types of colony cell growth develop, i.e., holo-, mero- and paraclones. They represent the proliferative compartment of human squamous epithelia. However, only the holoclone-forming cells possess full self-renewing capabilities and long-term regenerative potential, harbouring the features of epidermal SC. Notably, the term holoclone only describes the proliferative capacity of a keratinocyte in vitro. Nevertheless the progeny of a single epidermal holoclone can regenerate a fully functional epidermis in vivo. Their decendents, i.e., meroclone- and paraclone-forming cells, instead show a gradual loss of SC function with only limited proliferative capacity and self-renewal. Paraclone-forming cells are defined by a short replicative lifespan (up to 15 cell generations) after which they terminally differentiate, whereas meroclone-forming cells represent a transitional stage between the holoclone and the paraclone. The latter posses proliferative properties expected from transient-amplifying cells, which are an undifferentiated population in transition between SCs and differentiated cells2526.
The process by which adult epidermal SCs renew themselves and yield daughter cells depend on the tissue type and various other conditions, to include developmental stage, environmental injury, steady tissue turn-over and remodeling. Two models of epidermal differentiation and regeneration (hierarchical versus stochastical) have been described, in order to elucidate the nature and behaviour of interfollicular epidermal SCs, lying within the basal layer. The conceptual framework for these SC niches, their structure, compositions and operating process is steadily being updated (Fig. 2)27282930.
Stem cell niches
Skin SCs reside in specialized morphological and functional units with a specific microenvironment. These so-called niches may contain various SCs as well as supportive cells providing framework or signaling to the SCs31. Within human skin, at least five different niches have been delineated (basal layer of the epidermis, HF bulge, base of sebaceous gland, dermal papillae and dermis), that harbor different types of skin SCs32:
a) Interfollicular epidermal SCs are scattered singly across the dermal-epidermal junction. In the mucosa and on the palms and soles, SCs are located at the base of the rete ridges. They constitute about 1%~7% of epidermal basal cells. Several human SC markers have been described, including high surface expression of α6 and β1 integrins that may be relevant for sustaining the attachment of epidermal SC to their basement membrane through hemidesmosomes. Progenies from epidermal SCs that withdraw from the cell cycle, show a suppression of integrin α6 expression, before they start differentiating and moving towards the skin surface, where they slough off along terminal differentiation after approximately 4 weeks33. Furthermore, p63 (a homologue of tumor suppressor p53), a low expression of transferring receptor (CD71) and desmoglein 3 as well as LRIG1, the scaffold protein FERM domain-containing protein 4A (FRMD4A), and CD46 have been established as interfollicular SC markers3435.
b) Beside tissue regeneration interfollicular SCs have been shown to be invested with the ability of generating hairs32. In HFs, several distinct SC-types have been identified. One multipotent SC population resides in the bulge located at the base of the HF (during telogene phase of hair development) or beneath the HF-associated sebaceous gland (in anagen phase). This follicular component is established during embryonic hair morphogenesis and resists periodic degeneration during the hair growth cycle. Stimulation of these SC to exit their niche as well as their proliferation and differentiation to form mature HFs is closely linked to the hair growth cycle. HF bulge SC show expression of the molecular markers such as cluster of differentiation 200 (CD200), keratin 15 (K15), Lgr5+ and pleckstrin homology-like domain family A, member 1 (PHLDA1) as well as transcription factors Sox9+, Lhx2+ and NFATc1263637 Beside these epidermal SCs, another multipotent precursor cell population resides in HFs and dermal papillae that originate from the embryonic neural crest. These epidermal neural crest SCs (EPI-NCSCs) hold clonal multipotency that can give rise to melanocytic, neuronal and myogenic cell lineages in vitro and show differentiation potential toward mesenchymal lineages, as they are able to give rise to adipocyte, chondrocyte, and osteocyte progeny. Because of their advantageous physiological plasticity, multipotency, simple accessibility and non-controversial ethical issues, these EPI-NCSCs are considered promising donor cells for the repair of nervous system injuries38.
a) Sebaceous glands, attached to the HFs, are supposed to descend from different follicle SC populations, including Krt15+ bulge cells, LGR6+ and junctional zone SCs. Other studies describe the existence of periglandular Blump1-expressing sebaceous progenitors and a SC population within the gland itself. Progenitors give rise to terminally differentiated sebocytes that degenerate along holocrine secretion, releasing lipid-rich sebum into the hair canal that maintain an adequate lubrication of the skin surface39.
b) Melanocyte SCs derive from the neural crest and permanently reside in the HF bulge, basal epidermis and probably also in the dermis. They give rise to pigment-producing melanocytes in the epidermis and the hair matrix. The fate of the melanocytes within the follicle is connected to the HF phases, where melanocytes proliferate and differentiate during anagen, and diminish through apoptosis in catagen. Dysfunction of this SC population results in pigmentation defects that phenotypically manifest as hair graying. The latter underlies an increased apoptosis of melanocyte SCs due to higher oxidative stress subsequent to the deficiency of anti-apoptotic Bcl2 protein that occurs with aging1940.
c) The steady remodeling of the dermis and fibroblasts as their primary cellular component is managed via mesenchymal SCs. They are located in the connective tissue within the dermis, surround HFs (especially in the follicular sheath and papillae) or are found among pericytes around blood vessels. Beside fibroblasts, dermal mesenchymal SCs generate myofibroblasts, endothelial cells, nerves, blood vessels, osteoblasts, chondrocytes and adipocytes. Morevoer, they are crucial for the coordination of the complex process of wound healing by attracting other host cells, growth factors and extracellular matrix (ECM) secretory proteins. Dermal SCs lack uniform distinctive markers but adhere to plastic in contrary to other SCs41.
APPLICATIONS OF STEM CELLS FOR THE TREATMENT OF DERMATOLOGICAL DISEASES
General remarks on the therapeutic spectrum of skin stem cell populations
1) Epidermal stem cells (EpSC)
Advantages of using EpSC for research, diagnostic and therapeutic purposes include their readily accessibility and relatively simple isolation from (bioptic) skin tissues in comparison to ESCs. EpSC are further considered to be less “artificial” than iPSCs. Immune rejection following autologous transplantation is not expected and the tumorigenicity of these cells is considered to be low, due to their lesser degree of potency and absence of (epi-)genetic manipulations. In contrast, iPSCs reprogramming with tumorigenic c-Myc increases the frequency of transformed cells during iPSC generation. Tumor formation risk increases when the c-Myc transgene remains in established iPSCs and becomes reactivated42.
EpSCs demonstrate further favorable features, such as their high proliferation rate with the ability to double their number within 3~4 days of culture. At the same time they are able to keep their potency and differentiation potential for longer periods, although progressive aneuploidy (a state in which cells have abnormal numbers of chromosomes) and polyploidy (a state in which cells have one or more extra fully duplicated sets of chromosomes) as well as accumulation of mutations occur after several passages in cell culture. Notably, ethical issues do not restrict their use. This is in contrast to the serious ethical concerns that arise in ESCs research when referring to human dignity and ideas of personhood along the creation as well as destruction of embryos as the earliest forms of human life specifically for research purposes. All these characteristics make skin derived adult SCs an ideal population for the use in SC-based therapies2443.
Grafts generated from autologous epithelial cultures that encase an appropriate number of EpSCs as holoclones were shown to permanently recover massive epithelial defects (e.g., in skin and ocular burns or epidermolysis bullosa). Therewith, EpSCs also prove to provide both, a cellular environment and normal ECM to mediate restoration of a normal dermal-epidermal junction44454647.
2) Multipotent mesenchymal stromal cells (MSCs)
Since their first identification as fibroblast precursors in bone marrow in the 1950s, mesenchymal (stem) stromal cells (MSCs) have been obtained from several tissues, including adipose tissue, skin, umbilical cord blood, placenta, peripheral blood, endometrium, dental pulp, dermis, amniotic fluid, as well as from tumors4849. MSC of different origin share similar features but are not identical. Even in the skin several MSC subtypes exist. Regardless of their origin, MSCs possess a broad differentiating potential and some degree of plasticity, since they generate cells of not only mesodermal origin (i.e., osteocytes, adipocytes, chondrocytes, myoblasts, and tenocytes) but also of ectodermal (e.g., neurocytes, melanocytes) and endodermal lineages (e.g., hepatocytes, thyroid cells)50.
In 2006, the International Society for Cellular Therapy established guidelines for MSC characterization to counteract controversies concerning its name, definition, isolation and characterization criteria. The name “multipotent mesenchymal stromal cells” was favoured and three minimal criteria were delineated: i) adherence to plastic in culture; ii) expressing a combination of surface antigens (CD73+, CD90+, CD105+, CD34−, CD45−, CD11b−, CD14−, CD19−, CD79a− and HLA-DR−); and iii) in vitro differentiation-capability into adipocytes, osteoblasts and chondrocytes51. However, MSC populations isolated from different tissues significantly differ in their proliferation, differentiation and molecular phenotype.
Besides the differentiation-capability in vitro, the trophic, paracrine and immunomodulatory functions of MSCs are those that hitherto may have the biggest therapeutic implication in vivo6525354.
One of the main functions of MSCs is to support repair of damaged tissues. In response to inflammation MSCs migrate towards injured sites, differentiate into cells (mainly fibroblasts) and operate through the release of molecules participating in tissue regeneration such as cytokines (i.e., PGE2, GM-CSF, interleukin [IL]-1, RA, IL-7, IL-8, IL-10, and IL-11), growth factors and chemokines. In addition, MSCs modify tissue healing through pro-angiogenic, anti-fibrotic, and anti-apoptotic pathways. In stromal vascularized tissues, their perivascular amount correlates with the blood vessel density and the number of pericytes as mesenchymal progeny5055.
The immunomodulatory abilities of MSCs reside on the secretion of anti-inflammatory cytokines and the inhibition of CD4+ and CD8+ T cell, B-cell, and natural killer (NK) cell proliferation. These features depend on the microenvironmental milieu that MSCs encounter after their application. Thus, MSCs have been shown to exert even opposite effects in response to different inflammatory cues5056.
Although it is not fully determined whether MSCs are immunoprivileged or immunoevasive, they are specified as hypo-immunogenic due to their menial expression of major histocompatibility complex (MHC) class I molecules, as well as lack of MHC class II and co-stimulatory molecules, inclusive CD80, CD86, and CD40. These characteristics reduce the risk of immune rejection so that MSCs are considered to be safe when used in an allogeneic environment575859.
Up to date more than 600 clinical trials are listed in the U. S. National Institutes of Health database (www.clinicaltrials. gov) dealing with MSC therapy for various diseases such as different forms of cancer, spinal cord injury, multiple sclerosis, Parkinson's disease, myocardial infarction, rheumatoid arthritis and graft-versus-host-disease (GVHD)60. Current research strategies try to increase the efficiency of therapeutically administered MSCs by mainly encountering their limited persistence in vivo5059. Various methods have been applied to generate “optimized” MSCs, including genetic modification through viral and non-viral modifications, bioengineering of surface receptors, and priming with biological agents. For example, MSCs activated by nucleotide oligomerization domain 2 (NOD2; involved in the regulation of differentiation of umbilical cord derived MSCs and able to modulate inflammatory responses) or MSCs overexpressing SOD3 (a powerful antioxidant molecule) have been shown to exert much higher therapeutic efficacy than naïve MSCs in experimental immune modulatory models of atopic eczema and psoriasis, respectively. Although a confirmation of these results in the clinic is still missing, development of exceedingly efficient MSCs with augmented benefit and minimum risk along genetic modifications gives promising therapeutic perspectives5961.
3) Bone marrow stem cells
Bone marrow comprises at least two different lineages of cells: hematopoietic and associated supporting stroma with mesenchymal cells. Hematopoietic cells are produced by hematopoietic stem cells (HSCs), which are situated in the bone marrow SC niche. The mesenchymal compartment contains a subset of cells (1 in 107 to 108) with probably (pluri-)multipotent differentiation capacity, referred to as MSCs.
4) Bone marrow derived mesenchymal stromal cells (BM-MSCs)
The BM-MSCs are similar but somewhat different to mesenchymal stromal cells isolated from other tissues. The former can be isolated, enriched and transfused into allogeneic or autologous recipients along bone marrow transplantation (BMT) and exert a substantial role in producing erythrocytes, leukocytes, and platelets. They show also plasticity with their ability to differentiate into tissues of mesodermal, endodermal, and ectodermal origin, including skin626364 and have been implicated to contribute to skin development65. Nevertheless, the nature and function of these cells is still beeing controversial discussed.
BMT has been introduced as a kind of SC therapy over four decades ago. Although the initial implementation were for leukemia, lymphoma, hemoglobinopathies and aplastic anemia, the medical indications for BMT have extended and roughly 50,000 people worldwide receive this form of therapy annually63. Nevertheless, safety issues limit the application of BMT, since high morbidity and significant mortality is associated with BMT, even aggravated in patients with increased susceptibility to infections and a higher risk of tumor development66.
This therapeutic application has increasing relevance for skin diseases since several studies have reported that the BM poses a source of fibroblast-like cells in the dermis (along hematopoietic and mesenchymal lineages) and that their number augments in skin after wounding6768.
In addition, BM SCs may also serve as a reservoir for skin epithelial cells67. After BMT, donor cells differentiating into keratinocytes were detected in human epidermis of recipients for at least 3 years before vanishing. However, such BM-derived keratinocytes seem to be an extremely rare finding, perhaps contributing to only ~0.0001%~0.0003% of all epidermal cells in this setting69. Since BM-derived epithelial cells are sparse, the physiological role of BM cells in regeneration of the skin has been called into question.
Potential drawbacks of BM-MSC therapy refer to immune modulating abilities in context of a tumor microenvironment leading to an unfavourably alteration of anti-tumor response and angiogenesis. Furthermor MSCs may serve as precursors of tumor-associated fibroblasts and possess the capability to skew neutrophils and inflammatory monocytes or tissue macrophages into an immunosuppressive and tumor-promoting phenotype2070.
5) Induced pluripotent stem cells
Reprogramming of somatic cells to iPSCs provides an important (and ex vivo infinitely expandable) cell source to develop customized, patient-specific cells with a broad spectrum of cellular phenotypes for potential therapeutic applications7.
Skin cells like dermal fibroblasts, keratinocytes, dermal papilla cells or melanocytes are preferentially used for this technique, since they are easily accessible in the patient via isolation from punch biopsies. Especially fibroblasts further have plain culture conditions. Adult adipose SCs, yielded via lipoaspiration, pose another source for iPSCs71. The differentiation of both, mouse and human iPSCs into keratinocytes72, melanocytes73, and fibroblasts74 has already been successfully shown. This thus opens the possibility of extending iPSC technology into the field of dermatology17.
Interestingly, fibroblasts differentiated from iPSCs may display specific properties that exceed those of the parental fibroblasts from which these iPSCs were originally reprogrammed, such as an increased production and assembly of ECM75. Acquisition of an augmented biological potency of modified cells when compared to their parental origin is probably related to a modified epigenetic signature following differentiation of iPSCs and is an important functional feature for using these cells in regenerative therapies.
Fibroblasts are essential in maintaining normal tissue homeostasis and wound repair through their synthesis of ECM proteins and secretion of growth factors. Their incorporation into tissue-engineered biomaterials seems promising for the use in repairing damaged or diseased tissues by fabricating dermal substitutes74. In this context, iPSC-derived fibroblasts offer a novel source of autologous cells for dermal regeneration.
Although iPSCs have enormous potential for cell-based drug designs, cell therapy, and disease modeling, their transition into the clinic is still hindered by the missing evidence of safety and reliability of the reprogramming technology.
Although cell identity can be modified by the exogenous expression of transcription factors, the efficiency of nuclear reprogramming remains low (0.1% to 3%). This low outcome is probably associated with residual epigenetic memory of the tissue from which iPSCs were derived, detected via gene profiling studies in iPSCs. It is known that differentiated somatic cells have distinctive epigenetic patterns to maintain their cell identity. Cellular reprogramming works to change this epigenetic status of differentiated cells back to an undifferentiated state. Further, there is evidence that through the reprogramming process a restructuring of the existing somatic epigenetic memory takes place, followed by the generation of a new “epigenetic signature” adapted to the type of cell to be differentiated76777879.
In addition, currently available cell purification technologies may not fully succeed in separating the differentiated cells from undifferentiated iPSCs. Undifferentiated or partly differentiated iPSC could consequently be transplanted into the patient, carrying an increased risk of tumor/teratoma formation80.
Furthermore, it remains unclear to what extent the reprogramming process affects the genomic integrity of a cell. Several recent genomic analyses have signified that genomic abnormalities such as the accumulation of mutations and aberrant DNA methylation of distinct single bases emerge in iPSCs, either by the reprogramming process or following culture conditions8182. To address this issue, genome integration-free approaches are already widely used aiming at the reduction of the tumorigenic risk of insertion mutagenesis8384. However, it is necessary to perform more extensive and thorough genomic and epigenetic studies before using iPSCs in the clinic.
Interestingly not only iPSCs but also dermal fibroblasts themselves were demonstrated to have features of in vitro pluripotency without the necessity to be reprogrammed back to immaturity via activation of embryonic stage genes. Canadian researchers yielded a hematopoietic progenitor cell from a fibroblast through the in vitro implementation of specific cytokines. Consequently this hematopoietic precursor cell was able to give rise to granulocytic, monocytic, megakaryocytic and erythroid lineages, besides exhibiting the capability to repopulate the bone marrow by grafting85.
A recent study shows that terminally differentiated cells descending from mouse iPSCs do not provoke an immune response in syngeneic recipients86. This suggests that iPSC-derived cells might be well tolerated by the immune system. Again, more studies will be needed to definitively exclude a iPSC-mediated immune response in patients.
Immune rejection related to iPSC-based genetic correction is another problematic aspect, especially in skin diseases with homozygous null mutations of relevant genes. If a protein, that is unfamiliar to the host, is reintroduced, it may provoke an immune response and rejection of corrected iPSC-derived cells. This problem might be mitigated via prescreening for patients with compound heterozygous mutations or expression of nonfunctional, truncated variants of the protein of interest8087.
Stem cell therapy to enhance wound healing in the skin
Wound healing is a complex, dynamic process whereby the skin attempts to repair itself after injury. The delicate, coordinated wound repair process can be broadly divided into three phases (inflammation, proliferation and maturation) which all are susceptible to multiple interference factors that may result in chronic wounding88. In this context, initial approaches of SC-based wound therapies give a promising perspective50.
1) Epidermal stem cells (EpSCs)
EpSCs are a convenient target to use in wound therapies because they already reside within the skin and participate in the normal healing response. They have been shown to support healing by increasing proliferation and migration of fibroblasts and keratinocytes as well as enhancing angiogenesis by human vascular endothelial cells. In a type 2 diabetic nude mouse model, subcutaneous injection of allogeneic EpSC into acute full-thickness wounds have significantly shortened the healing time89. In a clinical study, a significant decline in wound size with increased reepithelialisation and vascularisation signs on histology taken after 4 months has been observed in 10 patients with non-healing leg wounds, after receiving autologous scalp-end terminal hair follicular grafts90. Terminal HFs (as a major SC niche) seem to play a decisive role in epidermal regeneration. This assumption is supported by a randomised controlled trial that compared the implantation of grafts containing scalp HFs with non-hairy skin grafts on chronic wounds in 12 patients. A significant reduction in wound size in the terminal HF-treated group was described9192. Notably, outer hair shafts diminish in most cases after a couple of weeks, hampering the development of HFs and subsequent cosmetic impairment by growing hair in engrafted transplants. Bulge SCs are highly relevant for epidermal renewal. In response to skin injury, these cells mobilize, leave their SC niche and contribute to repopulation of the epidermis while their progenies behave as transient amplifying cells with short lifespans37.
2) Multipotent mesenchymal stromal cell (MSC) therapy
MSC therapy is another emerging option to treat acute and chronic non-healing wounds. Beneficial effects are accomplished through structural repair via cellular differentiation, immunomodulatory responses, direct secretion of growth factors, advanced neovascularization and reepithelialization, as well as mobilization of resident SCs. Thereby, MSC play a pivotal role in all three healing phases. At the wound margins they stimulate the formation of granulation tissue by enhancing epidermal cell proliferation and growth of new blood capillaries. Further, endothelial cell recruitment is stimulated through the release of pro-angiogenic factors and growth factors such as vascular endothelial growth factor and angiopoietin-1. MSCs modify tumor necrosis factor-α production and lower NK cell function in the inflammatory phase, thereby reducing interferon-γ activity. In the last healing phase, scar formation is reduced through PGE2 secretion and lowering of TGF-β1 to TGF-β3 ratio, IL-10 up-regulation as well as IL-6 and IL-8 down-regulation. These effects are accompanied by a decline in collagen production and fibrosis5093. In addition, MSCs exert also antimicrobial activity via secretion of antimicrobial proteins or immune-modulating factors509495.
Several studies have already confirmed the positive effects of MSC (in particular MSC isolated from the skin, fat and bone marrow) on healing of acute and chronic wounds via induction and acceleration of regenerative processes50. Preclinical data demonstrated that local injection of BM-MSCs into an incisional full-thickness wound significantly shortens the healing time while stimulating angiogenesis, reepithelialization, and granulation96. Accelerated wound healing of diabetic ulcers has been also shown in preclinical and early human trials when BM-MSCs were used919497.
In an additional clinical study, autologous BM-MSCs were topically delivered via a fibrin spray to acute surgical wounds and chronic lower extremity wounds98. When assessed 20 weeks later, an accelerated healing of acute wounds and a significant reduction in size or complete healing of chronic wounds was observed. Efficacy of autologous BM-MSCs was further provided in the treatment of chronic non-healing ulcers of the lower extremities (diabetic foot ulcers, Buerger disease), reflected by a significant decrease in ulcer size compared to standard wound care99.
Several studies investigated the wound-healing effect of human adipose-derived mesenchymal stem cells (AD-MSCs) both in vitro and in vivo. AD-MSCs are clinically attractive as they can be easily isolated in relatively high quantity and possess satisfactory recovery potency100. In addition, it has been shown that the proliferative phase of healing involves the repopulation of adipocytes within skin wounds. Likewise, an in vivo mouse study suggested that immature adipocytes are activated during the proliferative phase alongside with mature adipocytes and fibroblast migration. Interestingly, lipoatrophic mice display impaired wound healing in comparison to controls, supposing adipocytes to be key elements of the intercellular communication during wound repair that mediate fibroblast migration and function101. The wound-healing effect of AD-MSCs by reducing wound size and accelerating reepithelialisation has been confirmed in several studies102103.
However, limitations to this data include short durations of in vivo experiments, small sample sizes, short follow-up periods, lack of randomized control trials and the use of animal model, as it is not always possible to directly extrapolate findings to the human wound physiology104.
3) Cell- and collagen-derived dermal scaffolds
Because the delivery of MSCs through direct injection or topically through gel matrices is detrimental for cell survival and usually causes significant rapid cell death, new strategies have been developed to improve MSC cell adhesion, proliferation and migration. These techniques are based on the use of MSC-seeded micro-or nanostructured scaffolds with natural biomaterials, such as collagen and cellulose derivatives9394105106. Thereby, pronounced to complete regeneration of non-healing wounds (burns, decubitus ulcers, diabetic ulcers) has been reached in preclinical and clinical studies107108.
For example, electrospun nanofibers of collagen and poly (d,l)-lactic-co-glycolic acid (PLGA) containing BM-MSCs were used to mimick the multilayer structure of the skin in the treatment of full-thickness skin wounds109. While the electrospun scaffold (i.e., highly porous) mesh constructs (generated by electospinning techniques using high-voltage electrostatic fields) provided mechanical support and protection of the wounds against external stresses, the hydrogel offered a physiological environment for MSCs proliferation. After implantation of collagen-PLGA scaffolds in vivo, MSCs promoted collagen synthesis and reepithelialization of the injured skin, while the biomaterial scaffolds were slowly degraded93110.
Stem cell therapies directed at autoimmune and inflammatory skin disease
MSCs have been employed with largely beneficial outcomes in the treatment of several autoimmune and inflammatory skin diseases particularly unresponsive to conventional therapy, to include acute and chronic GvHD with skin manifestations111, systemic lupus erythematosus112 and severe generalized systemic sclerosis (SSc)59. A number of further studies have demonstrated efficiency of MSC-based therapies for allergic immune disorders, like allergic rhinitis, atopic dermatitis and asthma. This data suggests that MSCs exert consistent anti-inflammatory/immune modulatory activities and are effective against disease-specific inflammation59.
The large majority of studies in this field used intravenous administration of allogeneic BM-SCs as the primary regimen to examine the effects on cutaneous inflammation and disease severity. In patients with severe psoriasis, allogeneic or autologous BMT was applied and showed clinical improvement. However, the risk of developing secondary autoimmune diseases, such as thyroiditis, autoimmune cytopenias, ulcerative colitis, systemic lupus erythematodes, insulin-dependent diabetes mellitus and myasthenia gravis was also increased. A switch in the T-cell subpopulations with suppression or delayed recovery of Tregs and elevation of Th17 as well as consecutive formation of autoantibodies due to a transient immune response imbalance during the reestablishment of normal immune and hematopoietic systems, were thought to underlie this phenomenon59.
It becomes increasingly manifest that MSCs from other tissues also possess immunomodulatory qualities similar to BM-MSCs, and that MSCs delivered via non-IV routes (i.e., topically, locally injection) can abate skin inflammation as well113114.
For example, subcutaneously administered human umbilical cord blood-derived MSCs (hUCB-MSCs) can effectively ameliorate the phenotype in an experimental mouse model of atopic dermatitis as well as imiquimod-induced psoriasis-like skin inflammation113115. In a clinical study, patients with moderate-to-severe atopic dermatitis benefitted from subcutaneous injection of allogeneic hUCB-MSCs, without developing considerable side effects116. There is one clinical trial (NCT02491658) currently underway, using hUC-MSCs intravenously for patients with moderate-to-severe psoriasis vulgaris. Preliminary results report on one patient that remained relapse free of psoriasis for five years after one dose of UCB-MSCs (1×106/kg), and a second proband who showed no symptoms of psoriasis for four years after receiving 3 infusions of UCB-MSCs (over 3 successive weeks) and 2 more infusions three months later117.
An additional case report from the Philippines investigated intravenous injection of autologous AD-MSCs in patients with psoriasis vulgaris. This study showed a significant improvement comparable to methotrexate treatment for mean 292 days without serious adverse events118.
Finally, transplantation using BM-MSCs in autoimmune diseases like lupus has resulted in significant clinical mitigation and functional amelioration of the affected organs119.
Given that no considerable adverse events have been reported in relation to MSC-based cell therapies, they seem to be safe and applicable for the treatment of severe, refractory immune-related diseases, especially for severely affected patients refractory to current first-line medications120121.
Lastly, comparing nonablative autologous transplantation of BM-SCs with the traditional treatment regimen comprising cyclophosphamide in a randomized study for SSc, the former therapeutic approach demonstrated reduction of skin and pulmonary symptoms for more than two years post transplantation, in comparison to conventional treatment122.
Stem cell therapy for epidermolysis bullosa
1) MSC-cell therapies
SC therapies are increasingly established in the experimental treatment of genetic diseases, recently also in patients with recessive dystrophic EB (RDEB), a rare genetic blistering disease. RDEB patients lack genes for synthesis of Collagen VII. These symptoms mainly account for more severe disease complications, such as mitten deformities of hands and feet and aggressive epithelial cancers.
Therapeutic approaches, either with intravenous infusion or direct local administration of MSCs to chronic wounds have started however, to provide novel insight into key BM cells and mechanisms germane to repair and regeneration of the epithelium.
Initially, intradermal injections of human BM-MSCs showed a dose-dependent, significant higher production and in loco deposition of type VII collagen associated with restoration of immature anchoring fibrils and superior dermal-epidermal integrity compared to controls with intradermal phosphate-buffered saline-injections in DEB mouse models123124125.
In line with this preclinical data, Conget et al.126 described the replenishment of type VII collagen at the dermal–epidermal junction upon intradermal BM-MSC injection provided from healthy donors into chronic wounds of two RDEB individuals. The administration led to a reduced blister formation (up to 6 months) and an increased reepithelialisation of chronic ulcers. Tissue remodeling activity of the transplanted MSCs, owing to both, their integration into the skin and their secretion of growth factors and cytokines participating in the regulation of tissue regeneration, might activate self-healing mechanisms in RDEB skin.
Subsequent studies questioned a predominant impact of SC on phenotypic amelioration. Petrof et al.127 showed that a single intradermal injection of allogeneic fibroblasts accelerated the initial rate of wound healing in patients with RDEB within the first 28 days although this effect diminished thereafter. In another study, both the injection of allogeneic cultured fibroblasts in suspension solution as well as of the suspension solution alone led to a similar increase in type VII collagen expression and improved wound healing in chronic non-healing ulcers of RDEB patients, independently of type VII collagen regeneration128.
The mechanical stimulus in the course of intradermal injection itself was thus supposed as a potential cause of improved wound healing, reflecting an elevated expression of heparin binding-EGF-like growth factor in response to subclinical inflammations that occur after injections into the human skin127.
Providing a clinically feasible approach of systemic treatment, also the infusion of allogeneic BM-derived SCs led to a phenotypic improvement in RDEB patients, showing reduced blistering and tissue fragility. Transplanted SCs from healthy donors are believed to engraft into the skin, differentiate to fibroblasts and keratinocytes, synthesize the missing type VII collagen anchoring fibers and thereby improve the fragile skin and shearing off of the epidermis63. In this respect, it has been shown that low levels of restored expressions of collagen VII (below 30%) are sufficient to significantly improve the RDEB phenotype and even a correction of about 3% COL7A1 mRNA seemed to be adequate for phenotypic reversion in a mouse model129130.
Ten RDEB children who received systemic (intravenous) allogeneic (wild-type) BM-MSCs showed improved wound healing, reduced skin redness, a subjective improvement in quality of life and high tolerability. As skin biopsies at 2 months post-treatment revealed no increase in type VII collagen nor new anchoring fibrils, phenotypic improvement possibly reflected a predominant immune suppressive and immune modulating effect of MSCs131. In another study with RDEB patients fewer blistering and significantly shortened healing time after treatment with BM-MSCs was demonstrated, either with or without concomitant cyclosporine132. In this cohort, electron microscopic examination additionally confirmed an increase in anchoring fibrils after treatment.
In summary, intradermal or intravenous injections of MSCs have shown some clinical benefits for RDEB patients. However, feasible application techniques as well as optimal cell dosage and frequency schedules for administration of allogeneic MSCs still have to be established and evaluated in future clinical trials. Further, biological and pratical limitations, like the ephemerality of the transplanted cells and potential immune rejection towards neo-antigens hitherto hinder the clinical applicability.
2) Bone-marrow transplantation
Following the demonstration of successful BM SC transplantation in murine RDEB, a clinical trial of whole BMT was performed in children with RDEB133. The first step included a high-dose chemotherapy to immunoablate patients to ensure more dependable lymphohematopoietic engraftment. Unfiltered cell populations of the donor BM were used based on previous observations that both hematopoietic and mesenchymal BM-derived SCs have the potential to increase the production of C7134. All six patients, who underwent BMT had some clinical improvement and five of the six showed increased C7 at the DEJ at the time biopsies where taken between day 100 and 200. Three of the six individuals showed an immense clinical improvement, with a reduction from 50% blistering area of the body surface to less than 10%. The remaining three patients exhibited a moderate improvement with less than 25% of the body surface area affected. However, toxicities occurred, as one patient died before the BMT due to heart failure possibly related to cyclophosphamide toxicity and pre-existing renal failure. Another individual died 6 months after transplantation because of an infection related to graft failure. Therefore, BMT protocols are currently refined by considering the use of reduced intensity conditioning and targeting of distinct subpopulations of BM cells implicated to be more effective, such as BM-derived Circulating PDGFRα+ mesenchymal cells)63134.
BMT has been also investigated in other forms of EB including severe generalized junctional epidermolysis bullosa (JEB)135. An important consideration in this form is, however, that the defective protein (laminin-332) is not synthesized by fibroblasts but keratinocytes. Thus, whether rectified donor BM-derived cells in the dermis are able/sufficient to regenerate the missing basement membrane protein, or a concurrent correction of epithelial cells is essential, still remains to be determined.
BM-derived MSCs were previously shown to differentiate into keratinocytes. Likewise, BM-derived hematopoietic and MSCs were demonstrated to be able to produce laminin- 332. Former clinical studies have further indicated that BMT increases laminin-332 expression at the dermoepidermal junction in some individuals136. Also other epithelial synthesized proteins involved in forms of EB associated with severe morbidity may be therapeutically targeted by BM-derived stem-cells. Thus, phenotypic improvement of type XVII collagen-deficient mice after BM-SC therapy suggests human studies in generalized intermediate junctional EB with deficient type XVII collagen63137138139.
3) Gene therapy
A low worldwide incidence of rare (or “orphan”) genetic skin diseases limits the accrual of data for research and suspends advancements in developing therapeutic concepts for these skin conditions. But since all of the most devastating forms of such inherited skin diseases like EB are mostly resulting from monogenic defects, in theory, their remediation at the genetic level should be more feasible. Sustained gene correction of continuously renewing tissue like skin, however, relies on the efficient molecular targeting of SCs.
A proof-of-principle study with somatic gene therapy in EB has been published44. Via a retroviral vector, a LAMB3 transgene has been inserted into autologous keratinocytes that were then grafted back as a confluent sheet onto the thigh of an adult patient with generalized intermediate junctional EB. In the following years, the graft has continued to express laminin-332 at the dermoepidermal junction, leading to a clinical improvement and long-term epidermal stability. This remarkable benefit was achieved although the number of holoclone SCs was reduced in the patient, probably a repercussion of long-term skin blistering resulting in niche destruction and SC depletion or exhaustion63140.
In the future, optimized protocols, with the goal to effectively isolate a sufficient amount of autologous EpSC, before being corrected by gene transfer, might facilitate the procedure. These cells, after building grafts and being transplanted, have the potential to induce long-term (if not permanent) regeneration of wounded skin. So far, promising observations of the use of genetically corrected skin grafts include (1) a total engraftment, yielding a morphologically and functionally normal, non-blistering skin that is able to resist mechanical stress, (2) continuing laminin beta-3-protein and laminin 332 expression that consequently strengthens the epidermal-dermal junction over a period of at least 8 years, (3) the ability to persistently and effectively restore the epidermis with only a few transgenic EpSC, which accounted only for a small subpopulation of transduced cells (most transduced keratinoctes have been shorter-living transit-amplifying progenitors)141.
Additional gene therapy studies, notably for RDEB and junctional EB, are currently at preclinical or early clinical stages. They mostly include keratinocyte grafting following ex vivo the introduction of a transgene or correction of both keratinocytes and fibroblasts with subsequent generation of a skin equivalent. Approaches to local gene therapy also include the injection of genetically corrected autologous fibroblasts harbouring the reparative transgene. Thereby potential pitfalls of wound infection (along the transplantation of skin equivalents) and graft loss (due to lower immunogenicity) in RDEB should be avoided. In contrast, intravenous gene therapeutic (systemic) approaches may be more feasible, tolerable and of systemic impact since the systemic nature of the disorder can be addressed63.
With regard to hitherto limited and/or transient efficacy as well as safety/tolerability issues, , optimal therapy for EB is doubtful to involve just allogeneic cells. More promise may hold a combination of gene, protein, drug, and cell therapies.
4) iPSCs
iPSCs therapies are therapeutically promising for genetic skin disease, because they can be rather easily exploited to be genetically manipulated. In addition, this approach makes modeling of skin diseases via targeted mutagenesis of the relevant genes possible forgoing the usage of ESCs142.
The successful establishment of iPSC-based therapies for hereditary skin diseases mainly relies on four steps: First, via a patient's skin biopsy cells have to be isolated. Second, these cells have to be transformed into iPSCs via genetic reprogramming. Third, genetic aberrations in obtained iPSCs have to be (safely) corrected, preferably through homologous recombination (HR). Fourth, these genetically corrected patient-specific iPSCs need to be differentiated into the desired cell type, followed by transplantation onto the same patient as an autograft.
Notably, even uncorrected iPSCs are valuable as disease- relevant patient-specific cells. They are used to customize in vitro disease models and in vivo xenograft models to gain new insights into disease mechanisms and drug discovery72.
Up to now, human iPSCs have been generated from several differentiated cell types derived from patients with certain genetic skin disorders. Besides type VII collagen (Col7)-deficient RDEB143144, LAMB3 gene-deficient JEB145 and EB simplex (EBS) with a dominant R125C keratin 14 mutation, these include, for instance, p63 mutant ectodactyly, ectodermal dysplasia, and cleft lip/palate syndrome, epidermolytic hyperkeratosis with a dominant N188S keratin 1 mutation, and dyskeratosis congenita, a multisystemic telomere disorder with several pathogeneic gene mutations146147.
In cell culture models, successfully generated iPSc from either gene corrected (autologous) RDEB fibroblasts or healthy (allogenic) individuals are able to differentiate into hematopoietic SCs and MSCs that can home to mucocutaneous blistering areas where they differentiate into keratinocytes and fibroblasts69143. Additionally, autologous grafting of in vitro generated 3-dimensional (3D) skin equivalents by iPSCs shows generation of stratified epidermis in vitro and in vivo (animal models)148149.
Up to now, several groups were able to show the therapeutic potential using corrected patient derived iPS cells in RDEB therapy studies in vitro17. In one study the correction of JEB-keratinocytes by iPSCs was done through a LAMB3-encoding lentivirus145. Another study used transfection with an expression plasmid encoding the wild-type human COL7 gene to yield a transient gene correction of COL7A1 gene-deficient RDEB-iPSCs144. The manipulated RDEB-iPSCs differentiated into structures resembling skin as well as cells of a hematopoietic lineage for BMT. In summary, corrected iPSCs have the potential to pose an unlimited source of autologous cells of both epidermal and mesenchymal lineages for the treatment of RDEB and perhaps other hereditary skin diseases150.
Nevertheless, this molecular approach harbours the risk of mutagenesis due to the application of viral and non-viral mediated gene correction and long-term cultivation involving multiple passages. Extensive genetic analysis (including whole genome/epigenomic assessment) will be necessary to purge the concern of off-target events that may occur after correction of iPSCs with techniques like ZFNs, TALENs and CRISPR/Cas systems. These drawbacks significantly impair the current clinical feasibility of iPSc-based strategies of gene correction17.
To overcome these obstacles, spontaneous reverted skin cells, so-called patient-specific naturally gene-reverted induced pluripotent stem cells, could be an alternative source of genetically corrected cells suitable for transplantation in patients with RDEB. This approach relies on the spontaneous reversions of inherited mutations that can be seen in different forms of EB, like RDEB, JEB, EBS (and many other genetic diseases and inherited skin disorders)151. Revertant mosaicism leads to somatic mosaicism that shows up as areas of normal skin in patients otherwise overwhelmingly affected by the disease-specific phenotype. This pattern is caused by a clonal outgrowth of cells harbouring acquired secondary mutations that reverse the effects of the primary mutation so that the phenotype locally diminishes. Several mechanisms are known to underlie this second “correcting” gene mutation in revertant cells, including gene conversion, second-site mutation and intragenic crossovers. Thus, if spontaneously revertant skin cells in EB are used to generate iPSCs, the risk of tumorigenesis via e.g. insertional mutagenesis after differentiation of these cells into hematopoietic (mesenchymal) or epidermal cells is reduced. Moreover, an unlimited source of naturally corrected cells for a cell replacement therapy could be obtainable151152.
The first attempt to use revertant cell therapy in an individual with generalized intermediate junctional EB yielded no functional benefits after (successful) grafting of isolated revertant keratinocytes, which were expanded to epidermal sheets. Of note, cultured keratinocytes showed 30% reversion, whereas the number of reverted keratinocytes dropped to 3% in the graft, probably because of lacking holoclones153. An alternative approach using punch graft transplantation of revertant skin, however, has been used successfully to heal chronic erosions with enhanced expression of laminin-332 in a patient with a similar form of junctional EB and mutated LAMB3 gene. The improved skin integrity was maintained for at least 18 months154.
The future impact of this approach thus relies on methods to more efficiently expand revertant keratinocytes in culture and to generate grafts containing adequate numbers of revertant SC to yield functional repair and regeneration of the skin.
Melanocytic diseases
McSCs are essential to maintain melanocyte populations in human skin and its appendages. Studies on McSCs have elucidated molecular mechanisms underlying ordinary melanocytic development as well as melanocyte-related pathological conditions like vitiligo and melanoma, although still many questions regarding the characterization of McSCs remain unsolved.
For example, it was traditionally assumed that cancer cells of melanoma arise from melanocytes. Recently, however, a hypothesis was posed that melanoma could also descend in extrafollicular SCs altered by harming factors such as ultraviolet (UV)A and UVB155. Experimental studies are currently ongoing to investigate the mechanisms capable of causing damage to the DNA of SCs, as to ascertain this hypothesis120. Human iPSCs may be useful for the characterization of human McSCs, since this application allows the acquirement of a sufficient amount of patient-specific melanocytes along the differentiation of iPSCs. These cells could then be applies for disease modeling and evaluation of potentially therapeutic approaches156. The use of HSCs transplantation, adjuvant to chemotherapy and immunotherapy for patients with metastatic melanoma, has been already evaluated in clinical trials. These strategies should allow the use of increased chemotherapy doses for more efficient eradication of tumor cells. However, definitive results are still missing120.
A distinct type of pluripotent, non-tumorigenic (in vivo) MSCs refer to the term multilineage-differentiating stress-enduring cells (Muse cells). These cells can be conveniently obtained from mesenchymal tissues (such as dermis and bone marrow) and human mesenchymal cultured cells (such as dermal fibroblasts). After culturing in a specific differentiation medium containing ten factors (Wnt3a, stem cell factor, endothelin-3, basic fibroblast growth factor, linoleic acid, cholera toxin, L-ascorbic acid, 12-O-tetradecanoyl-phorbol 13-acetate, insulin–transferrin–selenium, and dexamethasone), Muse cells derived from dermal fibroblasts have been shown to readily transform into functional melanocytes. These differentiated Muse cells expressed melanocytic markers, grew in 3D cultured skin and produced melanin after transplantation to the back skin of immunodeficient mice. However, in contrast to other pSCs such as ES cells and iPS cells, Muse cells show low telomerase activity and are not able to grow tumors in vivo. This technique might be the basis for new treatment approaches to melanocytic diseases like vitiligo157.
1) Cancer stem cells
Several years ago, it was discovered that a small sub-population of acute myeloid leukemia cells could reestablish tumors in severe combined immunodeficiency mice, while the vast majority of the tumor cells could not158. This study underlies the cancer SC hypothesis, which implicates that cancer SCs have characteristics comparable to the SC population of their tissue of origin (i.e., self-renewal, differentiation potential). They are assumable very rare within the tumor and are thought to produce progenitor cells that can generate all types of cells comprising the tumor. CSCs pose a challenge for cancer therapies, because eradicating the bulk tumor usually does not include all CSCs, leaving enough of them at liberty to re-establish the complete heterogeneity of cancer tissue.
In addition, these SCs might be more resistant to chemotherapy, and even targeted molecular therapies via their relatively high expression of the multi-drug resistance genes (e.g., MDR-1, BRCP1), a common feature of many SCs. Furthermore, owing to their ability to rapidly induce DNA repair mechanisms, CSCs are often highly resistant to radiation therapy. Finally, they appear to be particularly adept in stimulating angiogenesis, nurturing tumor development22.
CSCs can switch between quiescence (tumor dormancy) and active cell division with subsequent varying chemosensitivity, a behavior that mainly depends on changes in the microenvironmental niche and involves complex signaling pathways regulating tumorigenic growth and dormant arrest159.
CSCs further possess the capability to create new niches during the metastatic process160161. These “metastatic niches” are defined by specific locations (e.g., metastatic cells occupying native SC and perivascular niches), signaling pathways (e.g., PI3K-AKT pathway as a critical survival input for metastatic cancer cells), incorporated stromal (e.g., endothelial) cell types and ECM proteins (e.g., tenascin C which strongly promotes SC functions). The components of this micromilieu support the survival, self-renewal and expansion of disseminated metastatic CSCs.
Beside melanocytic SCs or melanoma cancer SCs probably involved in the pathogenesis of melanoma, CSCs have also been demonstrated in non-melanoma skin cancer such as squamous cell carcinoma (SCC) in mice. The proliferation and expansion of these CSCs are markedly influenced by their ability to respond to TGF-β receptor II and integrin/focal adhesion kinase-mediated signaling at the tumor–stroma interface. This pathway is crucially important in human cancer. It acts initially tumor suppressive (inhibition of proliferation) but promotes metastasis in later stages in response to a tumor associated altered cellular context and variable environmental signaling profiles162. Studies have revealed that several distinct CSC populations coexist in SCC and that tumor initiation and metastatic potential of these populations can be uncoupled. Therefor understanding CSC biology it is critical to develop novel CSC-targeted therapies, especially for patients with cancer and a poor prognosis163.
New therapeutics may be designed to specifically target these cells to block cancer progression. At the moment many chemotherapeutics attack rapidly dividing cells, so that it is easy for slowly dividing cancer SCs to evade these therapies40. Whether skin tumors like melanoma follow a cancer SC model for tumor development or a hierarchical model of tumor growth and progression (or combinatory/ other models) remains to be determined. These features of tumor dynamics, however, have implications on drug development in order to increase the efficacy of CSCs targeting19.
Cancer SCs from solid tumors usually express organ-specific markers. However, many caveats impede the discovery and identification of cancer SC markers for diagnostic and therapeutic purposes, to include the potential that the expression of these cell surface markers is not stable, that daughter cells may express different markers, that markers may not be unique to the cancer SCs but expressed in other cell types as well and that these surface protein markers may not have any role in cancer SC biology.
Beside the isolation of CSCs by flow cytometry according to CSC-specific cell surface markers, CSCs can be identified by so-called “side population chains (SP)” within a tumor. The latters refer to a subpopulation of tumor cells that is highly conserved in human cancer cell lines and linked to SC characteristics (clonogenic). It further features drug transport property with multidrug resistance and might serve as a “evolutionary backup” to keep alive at least a sub-fraction of cells when exposure to cytotoxic compounds occurs. SP show differential efflux activity to the main cell population usually measured by efflux of the fluorescent DNA binding dye Hoechst 3334. Moreover, CSCs may be determined through sphere assays, since tumorigenic cells showing SC characteristics have the ability to grow as floating spheres in serum-free medium19.
The significant role of aberrant Wnt signaling in cancer and CSC has engendered substantial efforts into the development of therapeutic approaches to target this pathway. Several small molecules, involved in tumor signaling, have been identified that selectively block the p300/β-catenin interaction, thereby increasing the CBP/β-catenin interaction, which maintains long-term pluripotency in a variety of SC populations. Thus the therapeutic potential of CBP/β-catenin antagonists (e.g., ICG-001) has been studied in various preclinical tumor models, where it has demonstrated the ability to safely eliminate drug-resistant tumor-initiating cells164.
Aging
Aging is a complex process which results from a multifactorial interaction of biological, biochemical and physical mechanisms that leads to structural and functional damage at both molecular and cellular levels with impaired (inter- and intracellular) signaling. SCs of the aged thus do not properly receive or respond to the normal, youthful, chemical and environmental signals that usually initiate normal differentiative SC responses. Notably, aging does not necessarily include an overall decline in the number of SCs, gene signatures, nor in their self-renewal capability. Instead, aging rather affects their function (i.e., differentiation and migration capability) which declines with age. Deficiencies or defaults in repairing the DNA, accumulation of toxic metabolites (e.g., reactive oxygen species), mitochondrial dysfunctions and epigenetic alterations have been suggested as underlying molecular mechanisms of impaired SC functions165165167.
The key mechanism of cell senescence and SC dysfunction during aging is believed to be oxidative damage. Excessive oxidative stress promotes expression of p53 and p16, molecules that trigger signaling pathways of cell apoptosis and premature senescence and probably inhibit directly or indirectly transcription factors that regulate (preserve) the ability to selfrenew and differentiate168.
There is an interesting relation of SC aging to hypoxia. Generally, SCs are maintained in a low oxygen environment or hypoxia in their native, quiescent state (5% of pO2 is the physiologically-relevant O2 concentration in their micro-environment). These conditions induce glycolysis in order to prevent oxidative stress-induced senescence. It has been shown that low oxygen atmosphere with 5% of O2 further promotes proliferation and maintains an undifferentiated state of umbilical cord MSCs in culture (with consecutively signs of rejuvenation)169. Since cellular aging goes along often with telomere length loss, it was observed that bone marrow MSC, expanded under hypoxia (3% O2) for 15 days, demonstrated telomere length maintenance. In contrast, telomere length decreased over time under normoxia (i.e., 20% O2), associated with a switch of MSCs to oxidative phosphorylation and a three to fourfold increase in senescense170171.
Additionally, also the micro-environment of SCs changes in the course of aging, with regard to the amount and composition of ECM, an altered expression of membrane proteins and lipids as well as changes in secretion of molecules. These alterations probably aggravate the signaling dysfunction, which gradually leads to a decline in homeostasis and tissue regeneration. Consistently, the preservation and rejuvenation of SC niches can reverse some phenotypic manifestations of aging 23172173.
With time SCs fail to keep the skin young-appearing. Consequently, normalizing the skin SCs activities could help reverse aged and photoaged skin, e.g. through generation of new fibroblasts, which subsequently produce new collagen, elastic fibers and further ECM substances. An increase in Wnt signaling has been associated with premature aging and decreased healing capability in mouse models. This might be related to a higher CBP/catenin interaction at the expense of the p300/catenin interaction, favouring symmetric divisions and quiescence. Therefor encouraging binding of b-catenin to its coactivator p300 in the nucleus or introducing small molecule CBP/catenin antagonists could provide a more optimal (youthful) balance in asymmetric versus symmetric divisions, so that a youth-like SC response might be restored. Since thousands of signals influence a SC's choice to proliferate or differentiate, supplying a single external/environmental signal or growth factor alone, however, might not be sufficient to restore proper SC function23167.
OUTLOOK
To date the preclinical and clinical studies on the use of SCs are exponentially increasing. Likewise, applications of SCs therapies are continuously expanding for various skin diseases.
Nevertheless, our current understanding of SC with regard to complex signaling cascades, environmental influences or epigenetic modulation is still limited. Further research is needed to improve the outcome and safety of SC therapies, reduce adverse events and identify new, potent targets for drug design.

 XML Download
XML Download