

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
X-linked agammaglobulinemia (XLA) is a primary immunodeficiency first characterized by Bruton1 in 1952. With an incidence of 1 in 100,000 to 1 in 200,000 of the population2, these individuals carry a mutation in the B-cell tyrosine kinase (BTK) gene encoding for a tyrosine kinase critical for B-cell maturation3. In myeloid and dendritic cells, BTK has been found to be a component of Toll-like receptor (TLR) signaling, important for recognition of foreign pathogens4. The coexistence of pyoderma gangrenosum (PG) in a patient with XLA has been rarely reported. PG is a neutrophilic dermatosis that occurs both as a primary disorder as well as secondary to an underlying disease. To date, only five previous reports have documented the association of PG with XLA56789. We report herein a case of PG in a patient with XLA.
CASE REPORT
An 8-year-old boy was transferred to our hospital for management of multiple, painful ulcers on the left lower extremity. The lesion began as a few small plaques on the left shin that gradually coalesced and broke down into an ulcer. Intravenous antibiotics were initiated but the ulcers continued to increase in size and number, which was associated with increased pain. There was no history of trauma to the lower extremities. He had been diagnosed with XLA at two years of age when he presented with recurrent pneumonia, pleural effusion and otitis media. He receives intravenous immunoglobulin replacement of every four weeks.
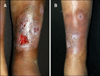
On clinical examination, the patient was febrile and had multiple ulcers with irregular, undermined, violaceous margins and indurated bases on his left shin (Fig. 1A). The right knee joint was swollen, warm, and tender with a detectable effusion, range of motion was limited by pain. There were no skin lesions on the trunk or genitalia. There was no associated lymphadenopathy.
Laboratory investigations showed that the patient had a normal white cell count, but a mild anemia (hemoglobin, 83 g/L; red blood cell, 4.8×109). Further laboratory data included an increased erythrocyte sedimentation rate (60 mm/h; normal range, 0∼15 mm/h) and C-reactive protein level (46 mg/L; normal range, 0∼8 mg/L). Immunological investigations were negative for antineutrophil cytoplasmic antibodies and antinuclear antibody. Blood and bone marrow cultures and virology screens gave negative results. Flow cytometric evaluation of peripheral blood lymphocytes showed markedly decreased numbers of CD19pos cells (0% of lymphocyte count), while other lymphocyte subsets were normal. The immunoglobulin (Ig)A and IgM levels were below normal (IgA <0.068 g/L [normal range, 0.51∼2.59 g/L], IgM <0.054 g/L [normal range, 0.48∼2.26 g/L]), whereas the IgG level was in the lower range of normal (normal range, 5.28∼21.9 g/L). The rest of the routine laboratory test results were within normal limits. During his hospitalization, the synovial fluid was negative for infectious organisms. Magnetic resonance imaging (MRI) demonstrated soft-tissue inflammation, and MRI revealed no evidence of osteomyelitis. The edge of the ulcer was biopsied and sent for histopathologic and microbiological cultures. The histopathological examination revealed a mixed neutrophilc and lymphocytic infiltrate and hemorrhage in the upper and lower dermis (Fig. 2). Special staining (Gram stain, periodic acid-Schiff stain) did not reveal any microorganisms. The culture of a biopsy specimen taken from the edge of the ulcer was negative for bacteria, fungi, and atypical mycobacteria. Based on distinctive clinical features, exclusion of infectious origin, and histopathologic findings, the diagnosis of PG was established. The patient received intravenous immunoglobulin treatment in conjunction with prednisone (1 mg/kg/d) and topical application of 0.03% tacrolimus ointment. On follow-up 2 weeks later, the ulcer was almost completely healed (Fig. 1B) and swelling of knee joint was relieved and no recurrence has appeared in the 2 months of follow-up.
DISCUSSION
We encountered a case of PG in a Bruton's XLA patient, in which defect in the BTK gene resulted in a decrease serum levels of immunoglobulin subtypes. However, there have been no reports that demonstrate an association between XLA and PG. Although the pathophysiology of PG is poorly understood, it is thought to be immune-mediated9, specially neutrophil chemotaxis and cytokine levels thought to be involved10. PG lesions either as a purely cutaneous manifestation or be associated with a systemic disease, including ulcerative colitis and Crohns disease, polyarthritis, IgA-gammopathy, malignancies and also with further conditions like chronic active hepatitis and Behcet syndrome11. To date, most patients have been described in single case reports, the association between PG and XLA needs larger sample sized studies supported obviously. Clinically PG starts with a sterile pustule, nodule or bulla that rapidly progresses and turns into painful ulceration with raised, undermined borders and necrotic eschar. PG-like ulcer caused by Helicobacter cinaedi and Campylobacter species have been reported in a patient with X-linked agammaglobulinaemia2. There was no clear difference about the morphologic characteristics of the mucocutaneous lesions between those and this case. In our patient, Tissue cultures were negative for infectious organisms.
The treatment of PG is still a therapeutic challenge. Initial therapy for PG consists of corticosteroids, dapsone, minocycline, methotrexate, cyclosporine, mycophenolate and intravenous immunoglobulin12. The majority of reported cases of XLA-associated PG required the use of immunosuppressive agents and immunoglobulin substitution for treatment. As BTK appears to be a component of TLR signaling, TLR activation leads to production of cytokines, notably tumor necrosis factor alpha (TNF-α), which promote the inflammatory cascade9. Therefore, anti-TNF agents, such as etanercept, have also been anecdotally reported to be successful in the treatment of PG13. Several reports has been demonstrated an improvement of cutaneous lesions of PG following topical treatment14. The combination use of intravenous immunoglobulin and prednisone and topical tacrolimus ointment showed great improvement in our patient. These cases suggest that treatment for PG should be individualized depending on the severity of the symptoms and underlying associated disease.

 XML Download
XML Download