

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor:
Hereditary epidermolysis bullosa (HEB) constitutes a diverse group of genodermatoses characterized by trauma-induced skin fragility, blisters and erosions. The fragility of the skin and mucous membranes results from abnormalities in the cytoskeleton of the basal keratinocytes or of the basement membrane zone1. Faced to the great heterogeneity of this disease, long and/or numerous exons of HEB genes, it was necessary to use next generation sequencing techniques, as an approach for disease-gene and variant causing identification2. In this study, we carried out whole exome sequencing (WES) analysis in one Libyan patient in order to identify the molecular aetiology of HEB.
The patient is the first child of a consanguineous healthy couple without a HEB familial history, originating from Libya. The baby was born without apparent abnormalities. After three days of birth, blisters followed by erosions and ulceration of skin sites prone to friction appeared. The mucous membranes were affected. Nail dystrophy was also observed. This study was conducted according to the declaration of Helsinki and to the ethical standards of the authors Institutional Review Board (Registration number IRB00005445, FWA00010074). After signing an informed consent, blood samples were collected and genomic DNA was isolated. WES was carried out using 5 µg of the genomic DNA sample.
Classical investigation of HEB is generally performed by electronic microscopy or/and immunofluorescence followed by mutational screening. However, this approach is expensive and time-consuming mainly in laboratories lacking logistics and human resources. Large genes like COL7A1 and PLEC1 and the presence of pseudogenes for KRT14 represent a major difficulty to perform molecular diagnosis. Direct sequencing of all COL7A1 coding regions might be challenging as they encompass 118 exons, and more than 70 polymerase chain reaction primer pairs are necessary for the amplification of all coding regions3. In addition, the genetic heterogeneity and the important number of “private mutation” make the genetic diagnosis more challenging even in endogamous populations4. WES has led to the identification of a considerable number of pathogenic mutations in monogenic Mendelian disorders, including skin pathologies5. Indeed, in comparison to individual genes such as COL7A1, Sanger sequencing costs are similar to the entire cost of WES3.
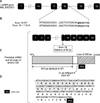
The library preparation, capture, sequencing, variant detection and annotation were performed by Oxford Gene Technology (OGT), (London, UK). Filtering steps were initiated by excluding reads with a coverage <20 and polymorphisms with a frequency >1%, according to OGT data, and polymorphisms for each gene involved in HEB were extracted (Table 1). Finally, all the variants reported in the Exome Variant Server and Exome Aggregation Consortium databases were excluded. These analyzes allowed the identification of a novel homozygous variant (c.2701+1G>A) occurring at the 18th donor splice site of LAMB3 gene in the affected sibling, and at the heterozygous state in the clinically unaffected parents. The c.2701+1G>A has been confirmed by Sanger sequencing of the corresponding DNA fragment. This variant was not present in 44 ethnically matched unrelated controls (88 chromosomes).
Splicing abnormalities are common in human diseases. These mutations can result in either complete skipping of one or more exons, retention of an intron or the introduction of a new splice site within an exon or an intron6. The most deleterious single nucleotide substitutions in donor or acceptor splice sites involve the +1/+2 or −1/−2 positions7. For our patient, the G to T transition at +1 bp position downstream exon 18 is predicted to abolish the 18th donor splice site according to Berkeley Drosophila Genome Project (http://www.fruitfly.org/seq_tools/splice.html). Thus, the affected splice site would lead to an aberrant transcript with an in-frame skipping of the last 82 nucleotides of exon 18. This aberrant splicing is predicted to result in a truncated β3 chain of laminin 332 with a loss of 25% (299 carboxyl-terminal amino acids) of the β3 chain due to a frame shift and a premature termination at codon 904. The predicted β3 chain mutant has 873 amino acids that are identical to the normal β3 chain while the last 31 amino acids are different (Fig. 1). The creation of a premature stop codon could result in the degradation of the LAMB3 mRNA by the nonsense-mediated decay mechanism.
The severity of the disease depends on the nature, the location and the impact of the mutation at transcriptional and translational levels. Mutations affecting the short arm of β3 chain of laminin 332 have been reported to be responsible of the Herlitz JEB (H-JEB) phenotype8. The H-JEB is characterized by profound skin fragility and mucocutaneous blistering with loss of fluids and proteins, making affected infants susceptible to infections. The long-term consequences include failure to thrive, anaemia, dyspnoea, pneumonia or sepsis, which explain the high mortality observed within the two first years of life9.
For these reasons and to avoid having other children with JEB, the family has requested a molecular diagnosis. By identifying the molecular aetiology of the disease, prenatal diagnosis has been planned to give parents the chance of having an unaffected child.
In this paper, we report on the molecular study of a Libyan patient with HEB. This is to our knowledge the first study of a Libyan patient. Our previous studies showed that despite mutational heterogeneity of HEB, some mutations are likely founder and closed to specific geographic regions4. Therefore, screening of c.2701+1G>A mutation could assist in determining the molecular aetiology of HEB among eastern Libyan patients.
Taken together, our results demonstrate that whole-exome sequencing could be used as a reliable and time-efficient technique for molecular aetiology determination, especially for heterogeneous disorders like HEB, particularly in unexplored areas.

 XML Download
XML Download