

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Degos disease is a rare systemic vaso-occlusive disorder. Degos-like lesions associated with systemic lupus erythematosus (SLE) are a type of vasculopathy. Almost all Degos-like lesions have the clinical pathognomonic appearance of porcelain-white, atrophic papules with peripheral telangiectatic erythema1. Such lesions can be found in at least two distinctive clinical settings: (1) apparent idiopathic disease, either classic Degos disease or its benign variant; and (2) cutaneous manifestations in several connective tissue diseases such as SLE, antiphospholipid syndrome, dermatomyositis, and systemic sclerosis2. The course, treatment, and prognosis of these diseases has substantially varied. To date, one case of systemic Degos disease with SLE has been reported in the Korean literature3. Herein, we report the first case of cutaneous Degos-like lesions without systemic involvement associated with SLE in Korea.
CASE REPORT
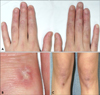
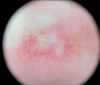
A 29-year-old woman presented to our clinic with a two-year history of asymptomatic atrophic white lesions. Two years ago, the lesions began as pink dome-shaped papules on her dorsal fingers and knees bilaterally. These papules gradually turned into umbilicated, atrophic porcelain-white lesions with a rim of erythema. Seven years ago, the patient was diagnosed with SLE and was being treated in a rheumatology clinic. On physical examination, atrophic, porcelain-white scars surrounded by an erythematous rim were seen on the dorsum of all fingers and both knees (Fig. 1). The patient also had a malar rash and arthritis, but did not have symptoms of gastrointestinal tract involvement or central nervous system (CNS) vasculitis, such as blurred vision and hemiparesis. Her neurological exam was unremarkable. On dermoscopy, a central whitish, structureless area surrounded by an erythematous rim of short vessels was seen (Fig. 2). Laboratory testing was positive for anti-nuclear antibody (1:1,280), anti-ds-DNA antibody, anti-smith antibody, anti-Ro antibody, and anti-La antibody, but negative for anticardiolipin immunoglobulin (Ig)G, anticardiolipin IgM, and lupus anticoagulant. Histologic findings showed hyperkeratosis, epidermal atrophy, and dermal sclerosis in the central portion. It also showed lymphocytic infiltration around vessels, fibrinoid necrosis of the vessel wall, and thrombus deposition in the lumen (Fig. 3). On the basis of these clinical, dermoscopic, laboratory, and histologic findings, the diagnosis of Degos-like lesions associated with SLE was made. The patient was being treated with hydroxychloroquine (300 mg/day), prednisolone (8 mg/day), and cyclosporine (50 mg/day) for seven years, and beraprost (20 µg/day) and pentoxifylline (400 mg/day) for 2 years for SLE. Because the patient's skin lesions improved gradually during the two-month follow-up period, we continued her treatment as before without further intervention, and no new lesions developed.
DISCUSSION
Degos disease, also known as malignant atrophic papulosis, is a rare systemic vaso-occlusive disorder first described by Köhlmeier1 in 1941 as erythematous papules with a central porcelain-white atrophic lesion. A combination of coagulopathy, endothelial cell damage, autoimmune disease, and vasculitis have been proposed as underlying pathogenic mechanisms, but none of these have been confirmed4.
Degos disease is classified into two types by associated clinical symptoms and underlying disease2. First, classic Degos disease has characteristic skin lesions and affects other organs with multiple limited infarcts. Skin lesions are usually the first manifestation of the disease, although its poor prognosis is the result of gastrointestinal and CNS involvement5. With systemic involvement, the reported mean survival time is approximately 2 years, but it varies from less than 1 year to more than 12 years6. Conversely, approximately 15% of Degos disease cases are a benign form often limited to the skin without gastrointestinal or CNS involvement. These cases generally exhibit long-term survival. Second, Degos-like lesions can occur in patients with connective tissue diseases such as SLE, antiphospholipid antibody syndrome, dermatomyositis, and systemic sclerosis78. Because of the broad overlap in clinical features and histopathological findings indistinguishable from those of cutaneous lupus erythematosus in skin lesions of Degos disease, Ball et al.8 did not conceive of Degos disease as a specific disease, but as a distinctive pattern expressive of different pathologic processes.
SLE is a chronic multiorgan auto-immune inflammatory disease and skin lesions are the second most frequent clinical manifestation. Nonspecific disease-related skin lesions are frequently seen, usually in the active phase of the disease9. Vasculopathy presenting as an acute or subacute manifestation of lupus can occur as Degos-like lesions and be etiologically implicated in the pathogenesis of Degos disease8. In our patient, there was no evidence of abnormality in platelet aggregation or blood fibrinolytic activity. She was found to have positive anti-nuclear antibody (1:1,280), anti-Ro antibody, anti-La antibody, anti-ds-DNA antibody, and anti-smith antibody titers at the time of diagnosis. Considering that the presence of vasculitis in patients with SLE is associated with anti-La antibody10, we assumed that our patient also had vasculitis, which can cause Degos-like skin lesions. By considering her clinical and laboratory findings, we suggested that her cutaneous Degos-like lesions were associated with SLE.
The skin lesions of Degos disease consist of three different evolution stages: (1) erythematous papules of onset; (2) erythematous papules with purple or necrotic centers with or without central crust; and (3) porcelain-white scars surrounded by an erythematous rim2. Dermoscopic findings consist of three different patterns, each corresponding to a different evolution stage. Dermoscopically, in the early stages, papules are characterized by a combination of a reddish-to-purple background and purpuric dots. At the intermediate stage, papules display a targetoid pattern with necrotic centers surrounded by erythematous halos. Finally, healed lesions have a whitish, structureless center surrounded by a rim of short, slightly curved vessels11. Harvell et al.12 described sequential changes in the histology of Degos lesions. Early lesions manifest a superficial and deep perivascular lymphocytic infiltrate, interstitial mucin, and vacuolar interface dermatitis. Fully developed lesions typically show prominent lymphocytic vasculitis, intramural fibrin deposition, central epidermal atrophy, and a developing zone of papillary dermal sclerosis and mucin deposition. Late stage lesions often demonstrate a classic inverted wedge-shaped zone of sclerosis with an atrophic epidermis and a sparse lymphocytic infiltrate in the upper half of the dermis. Given features of the three different stages, the cutaneous lesions in our patient were late stage, as all lesions corresponded with the clinical, dermoscopic, and histologic findings of that stage.
Currently, there is no consistently effective therapy for Degos disease, although Kim et al.13 reported that Degos-like lesions associated with Behçet's disease improved after treatment with aspirin and dipyridamole. Heparin has been successfully used in the acute phase, although other fibrinolytic agents have been ineffective. Immunosuppression with corticosteroids or other agents may worsen skin lesions and complications of Degos disease. Other therapies without proven benefits include antibiotics, arsenic, chloroquine, azathioprine, methotrexate, phenylbutazone, and interferon514. Our patient was treated with hydroxychloroquine, prednisolone, and cyclosporine for seven years, and with beraprost and pentoxifylline for two years for SLE. Her treatment was continued without modification, and her skin lesions slightly improved.
Considering all clinical, histological, and laboratory findings, we suggest that the cutaneous lesions in our patient were a reaction pattern. We suspect that her skin lesions were caused by peripheral arterial occlusion resulting from vasculitis associated with SLE. On the basis of our experience with this case, when pathognomonic Degos-like lesions that manifest with porcelain white scars surrounded by an erythematous rim occur, definitive diagnosis should be made after a careful clinical history and examinations that include physical, histological, and laboratory evaluation for SLE and other connective tissue diseases. Herein, we report a case of Degos-like lesions associated with SLE.

 XML Download
XML Download