

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Anaphylactic shock is a sudden, life-threatening allergic reaction associated with hypotension,1 following exposure to a specific antigen. An increased vascular permeability, one of the critical pathogenic factors for anaphylaxis, causes plasma extravasation, resulting in decreases in circulating blood volume and venous return, and finally systemic hypotension. In humans, within 10 minutes following onset of systemic anaphylaxis, circulating blood volume was lost by 35% due to increased fluid extravasation as evidenced by a considerable increase in hematocrit (Hct).2 Hemoconcentration is also observed in animals suffering from anaphylactic hypotension,345 and antigen-induced hyperpermeability has been well reported in the lesions of the lung, trachea, intestine, and skin. Limited studies have systematically examined the prevalence of increased vascular permeability among various organs affected. In rats, vascular permeability does not necessarily increase in a similar magnitude among organ affected, but the extent of vascular permeability changes vary considerably with the tissues: vascular permeability increases markedly in the trachea, moderately in the mesentery and intestines, and only slightly in the kidney, lung, heart, and liver.567 However, the regional differences in vascular permeability changes are not systematically reported in mice, the most frequently used experimental animals. Thus, the first purpose of this study was to determine the vascular permeability of various organs during systemic anaphylaxis in anesthetized mice sensitized with ovalbumin.
Patients with pre-existing cardiovascular diseases and those treated with propranolol, a β-adrenoceptor antagonist, have increased incidence and severity of anaphylaxis.89 Propranolol is a nonselective β-adrenoceptor antagonist and can inhibit both β1- and β2-adrenoceptors. We have recently demonstrated that the blockade of the β-adrenoceptor augments the severity of anaphylactic hypotension in anesthetized rats, and that inhibition of the β2-adrenoceptor rather than the β1-adrenoceptor exerts the detrimental action on systemic anaphylaxis via enhancing pulmonary vasoconstriction and bronchoconstriction.10 On the other hand, it is reported that inhibition of the β2-adrenoceptor augments anaphylaxis-induced vascular permeability in the skin and lung of rats.11 However, there have been few studies on modulating roles of β1- or β2-adrenoceptor in vascular permeability of various organs during anaphylactic shock in mice. Thus, the second purpose of the present study was to investigate the effects of β1- and β2-adrenoceptor antagonists on vascular permeability responses to anaphylaxis in mice.
MATERIALS AND METHODS
Animals
Eighty-seven male C57BL mice (Japan SLC, Shizuoka, Japan) weighing 27±1 g were used in this study. Mice were maintained at 23℃ and under pathogen-free conditions on a 12:12-hr dark/light cycle and were allowed food and water ad libitum. The experiments in the present study were conducted according to the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978) and approved by the Animal Research Committee of Kanazawa Medical University (2016-105).
Sensitization
Mice were actively sensitized by the subcutaneous injection of an emulsion made by mixing aluminum potassium sulfate adjuvant (2 mg) with 0.01 mg of ovalbumin (grade V; Sigma Chemical Co., St. Louis, MO, USA) dissolved in physiological saline (0.2 mL).12 The antigen emulsion was injected again 1 week after the first antigen injection. One week after the second injection, the mice were used for the following experiments.
Surgical preparation
Mice were anesthetized with intraperitoneal injections of medetomidine hydrochloride (0.75 mg/kg), midazolam (4 mg/kg), and butorphanol tartrate (5 mg/kg), and were placed supine on a heating pad (ATC-101B; Unique Medical, Tokyo, Japan) that maintained body temperature at 36–37℃. A polyethylene catheter (OD: 0.3 mm) was inserted into the left carotid artery for the measurement of mean arterial blood pressure (MBP). The right external jugular vein was catheterized using a 22-gauge polyethylene catheter (Terumo, Tokyo, Japan) (ID: 0.6 mm, OD: 0.9 mm). This catheter was connected to a Y-type miniature plastic tube, one twig end of which was connected via a water-filled polyethylene tube for the measurement of central venous pressure (CVP) and the other twig end was used to introduce a thin inner polyethylene tube, which was tapered to 0.1 mm in diameter over hot air for intravenous injections of the antigen, saline, and drugs.12
Experimental protocol
All experimental protocols were carried out in anesthetized mice. MBP and CVP were monitored using calibrated pressure transducers (TP-400T, Nihon-Kohden, Tokyo, Japan) positioned at the level of the left atrium. Heart rate (HR) was measured by triggering the wave of arterial blood pressure. The vascular pressures and HR were digitally recorded at 40 Hz by PowerLab (AD Instruments, Castle Hill, Australia).
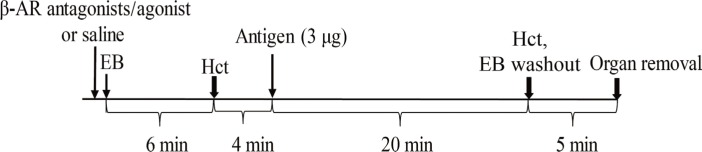
Fig. 1 shows the scheme of the time course of the experiments. The nonselective β-adrenoceptor antagonist propranolol (1 mg/kg; 100 µL; n=7), the selective β1-adrenoceptor antagonist atenolol (2 mg/kg; in 100 µL; n=7), the selective β2-adrenoceptor antagonist ICI 118,551 (0.5 mg/kg; 100 µL; n=7), or the selective β2-adrenoceptor agonist terbutaline (0.5 mg/kg; 100 µL; n=7) was intravenously injected at 10 minutes before intravenous injection of the antigen (3 µg) in 100 µL saline. Each β-adrenoceptor antagonist was also injected into the non-sensitized mice, as the β-adrenoceptor antagonist control groups (n=7/group). Saline alone (100 µL), as the vehicle of β-adrenoceptor antagonists or agonist, was injected into the sensitized control mice (n=7), non-sensitized control mice (n=7), and adrenalectomized mice (n=7). Immediately after injection of β-adrenoceptor antagonists, terbutaline, or the vehicle saline (100 µL), Evans blue dye (EB) was intravenously injected as described below. At 6 minutes after EB injection, the tail was cut with the stainless steel blade for sampling blood (50 µL) to measure baseline Hct. At 10 minutes, 3 µg of ovalbumin antigen in 100 µL saline was injected. The variables were measured for 20 minutes after antigen injection, followed by measurements of Hct and EB content of the tissues as described below.
All drugs were purchased from Sigma Chemical Company. All drugs were dissolved in saline. The doses of the antagonists were determined according to previous studies.131415 The dose of terbutaline was determined preliminarily so that systemic effects of this substance, such as hypotension and tachycardia, were minimum.
Adrenalectomy
To test the possible contribution of endogenous epinephrine to changes in vascular permeability, both adrenal glands were resected via a dorsal lateral incision by separating the gland from the surrounding tissue with tweezers and then gently pulling the gland. Then the incisions in the muscle walls and flank incision were closed with silk sutures.
Measurement of vascular permeability
Vascular permeability was measured by quantifying EB bound to albumin in various tissues, such as the trachea, lung, mesentery, small intestine, heart, kidney, spleen, adrenal gland, liver, and limb skeletal muscle. Mice received EB (27 mg/kg, 1%) immediately after injection of each antagonist and agonist or saline. At 20 minutes after injection of the antigen, mice were killed after injection of 0.1 mL pentobarbital and perfused with saline using a double heads Masterflex pump (Cole-Parmer, Vernon Hills, IL, USA) through the left and right ventricles to remove intravascular EB in the pulmonary and systemic vessels, respectively, at the rate of 10 mL/min for 5 minutes. The tissues were removed, weighted, and homogenized in 0.5 mL of formamide and incubated at 60℃ for 48 hours. Then, the supernatant was separated by centrifugation at 12,000 g for 30 minutes and was added on a clear 96-well plate (120 µL per well). The optical density of the supernatant was determined by spectrophotometry at 620 nm. The concentration of EB in the tissue was determined from the generated EB standard absorbance curves. Data are expressed as µg of EB/g of wet weight tissue.
Measurement of plasma catecholamine concentrations
To determine plasma concentrations of epinephrine and norepinephrine during systemic anaphylaxis, the sensitized control mice (n=5), adrenalectomized mice (n=5) and non-sensitized control mice (n=5) were used in separate experiments, in which MBP was also measured in the same way as described above. At 6 minutes after antigen administration, blood (0.6 mL) was sampled from the left carotid artery. Blood samples were transferred immediately to chilled tubes containing EDTA, and then centrifuged (12,000 g, 10 minutes, and 4℃). The plasma samples were separated and stored at -80℃. Plasma catecholamine concentrations were determined by high-performance liquid chromatography with trihydroxyindole reaction.
Statistical analysis
Results are expressed as means±SE. For the analysis of the variables, intragroup and between-group comparisons were made using one-way and two-way analysis of variance for repeated measures. When a significant difference was observed with the two-way analysis of variance, paired comparisons were made within a group and between groups by using the Dunnet and Tuckey posttest, respectively. Kaplan-Meier survival curves were analyzed using the log-rank test. Significance was assumed when a P value was less than 0.05 (two-tailed). All statistical analyses were performed using KaleidaGraph 4.0 (SAS Institute Inc., Cary, NC, USA).
RESULTS
Mouse anaphylactic response
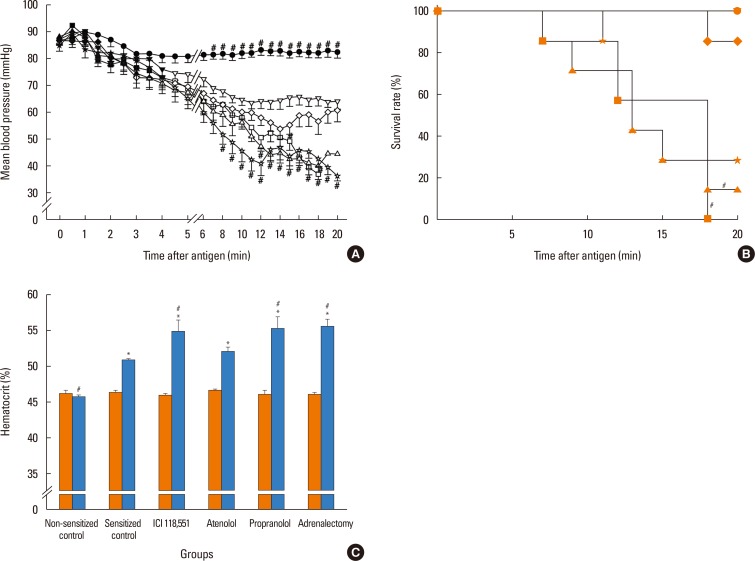
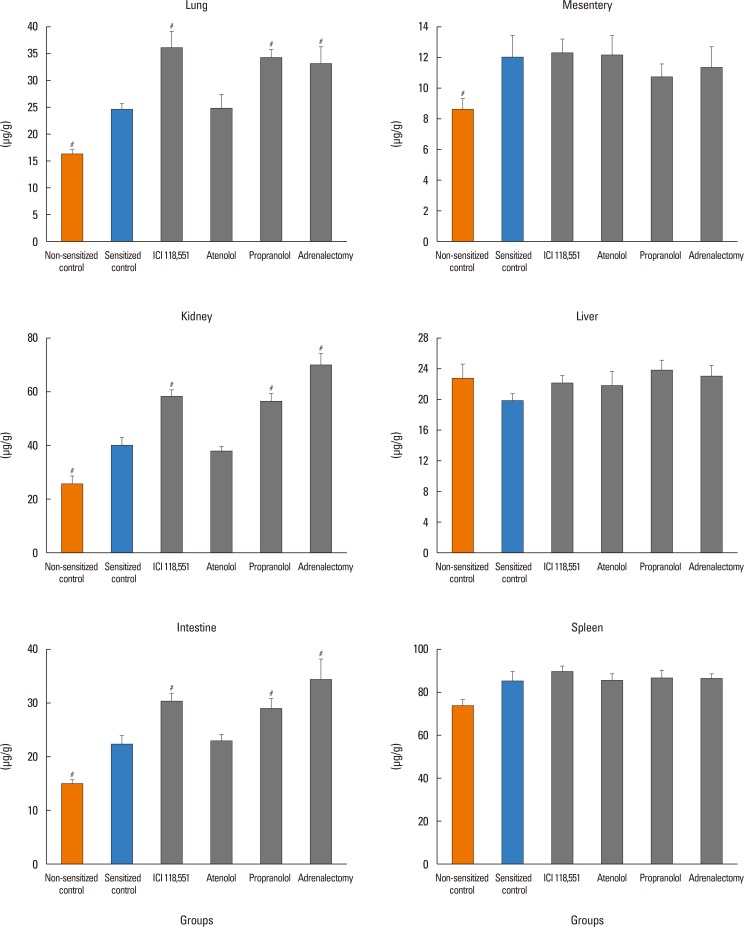
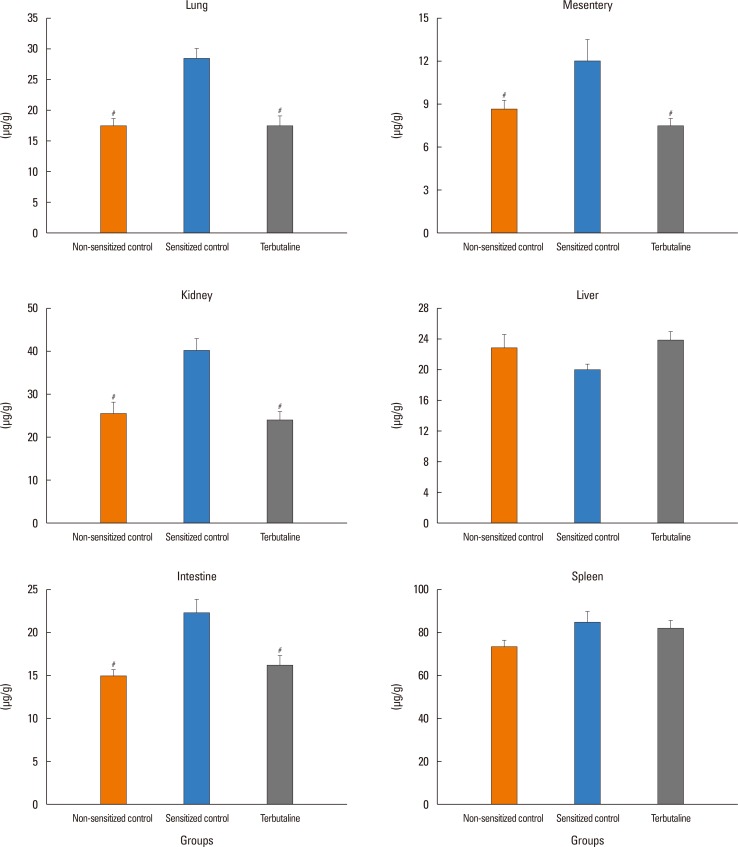
In the sensitized control group, MBP significantly decreased from the baseline of 87±4 to 64±7 mmHg at 20 minutes after antigen injection. CVP and HR significantly decreased and increased, respectively (data not shown). No mice injected with antigen died by 20 minutes, the end of the experimental period (Fig. 2B). Hct increased by 5%±1% (Fig. 2C). As shown in Fig. 3, EB content of the lung, kidney, mesentery, and intestine, but not the liver or spleen, were significantly greater in the sensitized control group than in the non-sensitized control group: EB extravasation was greater by 52% for the lung, by 57% for the kidney, by 50% for the intestine, and by 39% for the mesentery, in the sensitized control group than in the non-sensitized control group (Fig. 3). However, the EB content of the trachea, heart, adrenal gland, and limb skeletal muscle were not detected probably due to small sample sizes for the trachea and adrenal gland or low vascular permeability for the heart and limb skeletal muscle.
Effects of β-adrenoceptor antagonists, adrenalectomy and terbutaline
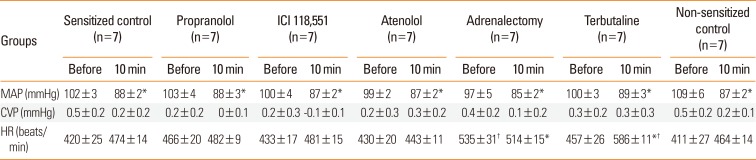
Table shows the effects of β-adrenoceptor antagonists, terbutaline, and adrenalectomy on the basal levels of the variables. MBP decreased at 10 minutes after injection of β-adrenoceptor antagonists or saline due to blood sampling (50 µL) for the measurement of baseline Hct at 6 minutes. There were no significant differences in MBP, HR, or CVP at 10 minutes, immediately before antigen injection, among the groups studied.
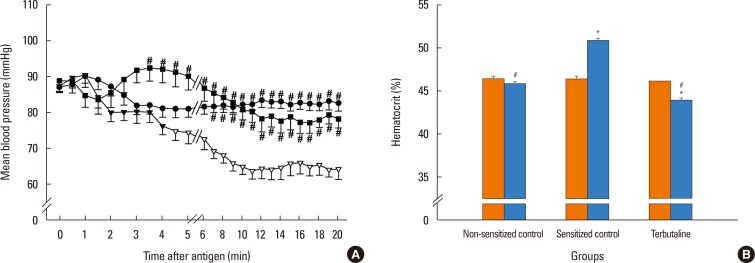
Pretreatment with propranolol, ICI 118,551, and adrenalectomy, but not atenolol, augmented the anaphylaxis. In mice pretreated with the propranolol, ICI 118,551, and adrenalectomy, the survival rate and Hct were smaller and greater, respectively, than in the non-pretreated sensitized control mice injected with antigen (Fig. 2B and C). MBP showed significantly a greater decrease in mice pretreated with propranolol, ICI 118,551, and adrenalectomy than in the sensitized control mice (Fig. 2A). Decreased CVP and increased HR were similarly observed in the sensitized mice after antigen injection (data not shown). As shown in Fig. 3, in mice pretreated with propranolol, ICI 118,551, or adrenalectomy, EB content in the lung, kidney and intestine, but not in the mesentery, liver or spleen, were significantly greater than those in the sensitized control mice: as compared with the EB content of the sensitized control group, those of the ICI group were significantly greater by 47% for the lung, by 47% for the kidney, and by 36% for the intestine; those in the propranolol group by 39% for the lung, by 41% for the kidney and by 30% for the intestine; those in the adrenalectomy group by 34% for the lung, by 75% for the kidney, and by 54% for the intestine (Fig. 3). In any β-adrenoceptor antagonist control groups, variables did not significantly change after antigen injection, and EB extravasation did not significantly differ from that in the non-sensitized control group (data not shown). These findings suggest that β2-adrenoceptor antagonist augments anaphylactic hyperpermeability and deteriorates systemic anaphylaxis. Therefore, we next examined whether β2-adrenoceptor agonists provide beneficial effects on anaphylaxis. As expected, terbutaline pretreatment abolished antigen-induced increase and decrease in vascular permeability and MBP, respectively, and rather decreased Hct after antigen injection (Figs. 4 and 5).
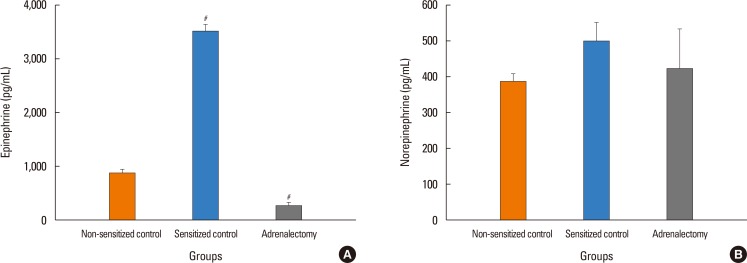
To clarify the possible roles of endogenous catecholamine in the attenuation of antigen-induced hyperpermeability, as described above, the plasma levels of epinephrine and norepinephrine were measured at 6 minutes after antigen challenge, when MBP fell, in the separate experiments. The plasma epinephrine levels of the sensitized control mice after antigen injection (3,524±391 pg/mL) were 4- and 13-fold greater than those of the non-sensitized control mice (869±206 pg/mL) and adrenalectomized mice (264±164 pg/mL) (Fig. 6A). There was no significant difference in norepinephrine levels between these 3 group mice (Fig. 6B).
DISCUSSION
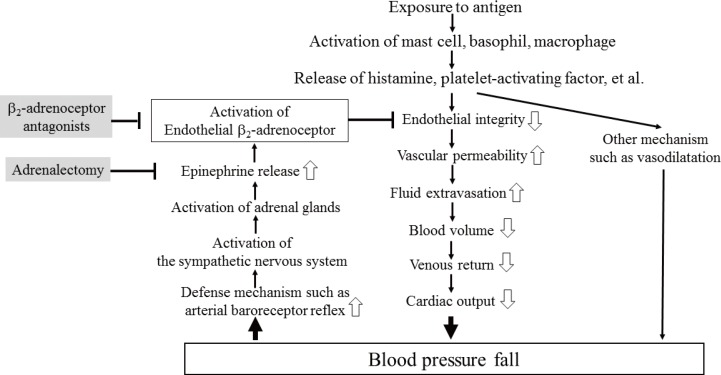
In the present study, we determined the effects of β-adrenoceptor antagonists and adrenalectomy on the responses of MBP, EB extravasation, Hct, and the survival rate during systemic anaphylaxis in anesthetized mice. There are 2 main findings in the present study. One is that, injections of the antigen into the sensitized mice caused systemic hypotension, increased vascular permeability, and hemoconcentration, which suggests that antigen-induced increases in vascular permeability cause fluid extravasation with resultant sequential decreases in circulating blood volume, venous return, and finally MBP; the other is that mice pretreated with propranolol, ICI 118,551 and adrenalectomy, but not atenolol, showed greater responses to anaphylaxis than the sensitized control mice. Furthermore, the plasma levels of epinephrine increased markedly after antigen in the sensitized mice, but not in the adrenalectomized mice. These findings clearly indicate that inhibition of epinephrine release from the adrenal glands and blockade of β2-adrenoceptor, rather than β1-adrenoceptor, augment the antigen-induced increase in vascular permeability and deteriorate anaphylactic hypotension. In other words, epinephrine endogenously released from the adrenal gland activates β2-adrenoceptor, resulting in attenuation of an anaphylaxis-induced increase in vascular permeability and hypotension. Moreover, we have reinforced the beneficial role of β2-adrenoceptor activation in mouse anaphylaxis by showing that the β2-adrenoceptor agonist terbutaline exogenously administered abolished anaphylactic alterations.
In the present study, we clearly showed that anaphylaxis increased vascular permeability by 39%–57% in the lung, intestine, kidney, and mesentery, but not in the spleen or liver (Fig. 3). The blood vessels in the spleen and liver are highly permeable because capillaries of these organs are lined with fenestrated endothelial cells.1617 Therefore, even though they are exposed to edematogenic agents, vascular permeability could not be furthermore enhanced. In contrast, the other organs examined have semipermeable continuous endothelial cells, the permeability of which could be increased by anaphylactic agents, such as histamine and platelet-activating factor (PAF). In the present study, the vascular permeability increased to a similar degree in the lung, kidney, intestine, and mesentery of mice during systemic anaphylaxis. This finding indicates that there are little differences in increased permeability among these organs during systemic anaphylaxis in mice. These results in mice contrast with those in rats: vascular permeability does not necessarily increase similarly among organs in rat anaphylaxis, with the permeability increasing markedly in the trachea, moderately in the mesentery and intestines, and slightly in the kidney, lung, heart and liver.567 In the present study, permeability changes in the trachea could not be evaluated due to too small-size samples. The reasons for the differences in leaky organs during anaphylaxis between rats and mice are not known, but may be ascribed to species differences in regional anaphylactic responses.
The mechanism for the anaphylaxis-induced increase in vascular permeability in the present study could be ascribed to chemical mediators released from mast cells, basophils and macrophage via anaphylactic reactions.18 We previously reported that PAF and histamine play major roles in anaphylactic hypotension of anesthetized mice.19 Indeed, histamine and PAF are well known to increase vascular permeability, although the exact mechanism remains unsettled.2021 On the other hand, vascular permeability is regulated by the maintenance of endothelial cell junctions. The specialized structures maintaining endothelial cell-to-cell adhesion are adherens junctions, in which vascular endothelial cadherin (VE-cadherin) forms zipper-like structures along endothelial cell contacts.2223 Dysfunction of adherens junctions leads to increased paracellular transport or vascular permeability through endothelial cell contraction-dependent and contraction-independent mechanisms.24 In this respect, histamine increases vascular permeability by adopting both mechanisms,252627 while PAF does by destabilizing VE-cadherin in a way independent of endothelial cell contraction.27
It has been shown that β-adrenoceptor-mediated signaling contributes to endothelial barrier maintenance under baseline conditions.2829 However, in the present study, β-adrenoceptor antagonists themselves did not affect changes in either Hct or EB extravasation of the mice without antigen injection. In contrast, blockade of the β2-adrenoceptor augmented the antigen-induced increase in vascular permeability in this study. The mechanism for the enhancing effects of the β2-adrenoceptor blockade on increased vascular permeability induced by the antigen may be related to intracellular cAMP: cAMP produced in endothelial cells by activation of β2-adrenoceptor via adenylate cyclase activation causes activation of protein kinase A, leading to inhibition of RhoA and then myosin light chain (MLC) kinases, resulting in decreases in MLC phosphorylation and finally endothelial contractility.30 Under cAMP/PKA-dependent stimulation, MLC phosphorylation was also reduced by activation of MLC phosphatase.31 In addition, cAMP activates Rac-1, which causes assembly of actomyosin proteins and enhances the integrity of endothelial cells.32 β2-adrenoceptor blockade apparently inhibits the development of these beneficial effects of cAMP. In agreement with this assumption, in the present study, β2-adrenoceptor activation by terbutaline abolished anaphylaxis-induced hyperpermeability.
In the present study, adrenalectomy also enhanced the anaphylaxis-induced increase in vascular permeability. In the basal states without β2-adrenoceptor blockade, epinephrine is released from the adrenal glands in response to anaphylactic hypotension, resulting in activation of the β2-adrenoceptors of endothelial cells that causes cAMP-dependent enhancement of endothelial integrity or decreased vascular permeability. In other words, β2-adrenoceptor might be intrinsically activated by epinephrine, with resultant attenuation of the anaphylaxis-induced increase in vascular permeability. Actually, we found that the plasma epinephrine levels increased 4-fold after antigen injection in the sensitized control mice, whereas they were considerably low in the adrenalectomized mice. This finding strongly suggests that epinephrine was released from the adrenal glands during anaphylaxis. In fact, we observed that efferent sympathetic nerve activity to the adrenal gland increased in mice suffering from anaphylactic hypotension (unpublished observations).
Concerning explanations for the deterioration of the survival of mice with anaphylaxis by β2-adrenoceptor inhibition in the present study, we assumed that the β2-adrenoceptor antagonist further increased vascular permeability, reducing effective circulating blood volume and then enhancing anaphylactic hypotension. Furthermore, β2-adrenoceptor antagonists may induce substantial coronary vasoconstriction of the heart,33 while activation of β2-adrenoceptors in the heart causes coronary vasodilatation.3435 Consistently, inhibition of cardiac β2-adrenoceptor enhanced anaphylaxis-induced coronary vasospasm, with resultant decreased cardiac contractility,36 which could reduce cardiac output and then augment anaphylactic hypotension. As the other mechanism, β2-adrenoceptor antagonists might exert detrimental effects on the antigen-induced pulmonary vasoconstriction and bronchoconstriction, as observed in the β2-adrenoceptor antagonist-pretreated rats.10
In summary, we determined vascular permeability responses to anaphylaxis in anesthetized C57BL mice, with special attention to the influences of the β1- and β2-adrenoceptor antagonists. The content of this study is summarized in Fig. 7. Injections of the ovalbumin antigen into the sensitized mice increased vascular permeability in the lung, intestine, kidney, and mesentery, but not in the spleen or liver. MBP and Hct were decreased and increased, respectively. In mice pretreated with propranolol, ICI 118,551, and adrenalectomy, the survival rate and Hct were smaller and greater, respectively than in the sensitized control mice injected with 3 µg antigen. The antigen-induced increase in vascular permeability in the kidney, intestine, and lung was augmented following the β2-adrenoceptor blockade and adrenalectomy. Furthermore, the β2-adrenoceptor agonist terbutaline abolished the antigen-induced alterations. The plasma epinephrine levels were significantly greater in the sensitize control mice than in the non-sensitized control mice during anaphylaxis. These findings clearly indicate that activation of β2-adrenoceptors by epinephrine endogenously released from the adrenal glands can attenuate an antigen-induced increase in vascular permeability in anesthetized mice.
 XML Download
XML Download