

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Asthma is a chronic inflammatory disorder affecting 300 million individuals worldwide.123 Symptoms of asthma, including episodic airway obstruction, airway hyperresponsiveness (AHR), and reduced lung function, are due to chronic airway inflammation and underlying structural changes in the airway wall.4567
The bronchial epithelium is constantly exposed to a wide range of environmental materials present in inhaled air, including noxious gases and anthropogenic/natural particulates, such as gas and particles from car emissions, tobacco smoke, pollens, animal dander, and pathogens.8 As a fully differentiated, pseudostratified mucociliary epithelium, the bronchial epithelium protects the internal milieu of the lung from these agents by forming a physical barrier involving adhesive complexes and a chemical barrier involving secretion of mucus, which traps inhaled particles that can be cleared by the mucociliary escalator.8 The bronchial epithelium as the initial cell in contact with the environment also plays a pivotal role in immune surveillance and appropriate activation of immune effector cells and antigen presenting cells in the presence of pathogens or other danger signals.8
The airway epithelium closely exposed to the external environment plays an important role as a physical barrier and a modulator of allergic response, and leads to allergic inflammation.29 Barrier dysfunction in the lung causes allergens to affect the epithelium and produce various cytokines that mediate airway inflammation.10111213 Epithelial barriers consist of airway surface liquids, mucus, and apical junctional complexes that form between neighboring cells.13
Tight junctions (TJs) act as a barrier to the paracellular transport of ions, solutes, and water, as well as cells, and function as a fence that divides apical and basolateral domains of plasma membranes.1415 TJ cleavage and repair occur following exposure of epithelial cells to allergens, suggesting that disruption of lung epithelium permeability barrier by allergens may be an important event in allergic sensitization and asthma.161718
Claudins are structural molecules of TJs, and different claudins (about 27 claudins) are responsible for changes in electrolyte and solute permeability in cell layers.19 Claudin-4 has been reported to function as a paracellular sodium barrier and is one of the 4 major claudins expressed in lung alveolar epithelial cells.2021 The role of claudin-4 is studied in lung injury,22 cancer,23 and fibrosis.24 However, the role of claudin-4 in the pathogenesis of asthma is not clear. Up to date, there have been few data about claudin-4 in asthma. Also, this study was to investigate the role of epithelial barrier claudin-4 in the pathogenesis of asthma. Moreover, we also determined claudin-4 levels in blood from asthmatic patients between stable and exacerbated states.
Go to : 
MATERIALS AND METHODS
Patients and Control Subjects
The biospecimens and clinical data were provided by the biobank of Soonchunhyang University Bucheon Hospital, a member of the Korea Biobank Network. All subjects had a clinical diagnosis of asthma according to the Global Initiative for Asthma (GINA) guidelines.2 All subjects underwent standardized assessments that included complete blood cell and differential counts, IgE measurement, chest posteroanterior radiography, allergy skin prick tests, and spirometry. All data were collected at the time of diagnosis, before administration of asthma medication. Exclusion criteria included respiratory infections within 4 weeks of screening, smoking history of >10 packs per year, chronic obstructive pulmonary disease, and parenchymal lung disease apparent on chest radiography.
Normal control subjects were recruited from the spouses of the subjects or members of the general population. Clinical data from patients and control subjects are presented in Table as previously described.25
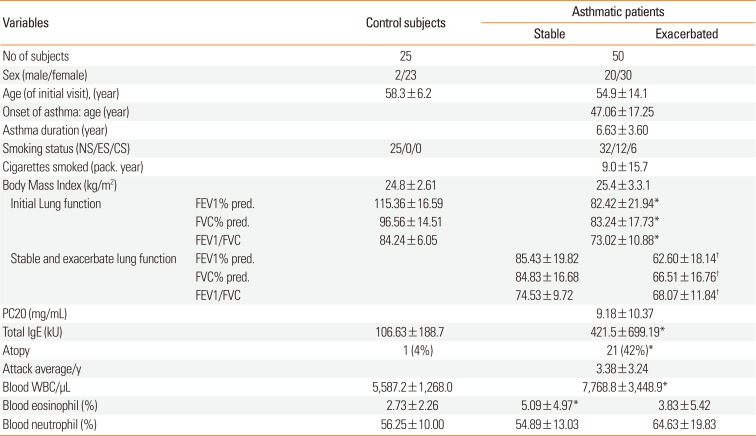
Table
Clinical characteristics in control subjects and patients with asthma
Data are expressed as mean±SD.
PC20, the concentration of methacholine required to decrease FEV1 by 20%; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; BMI, body mass index; SM, smoker; ES, ex-smoker; NS, non-smoker.
*P<0.01 compared with control subjects; †P<0.05 compared with stable asthmatics.
![]()
Asthma exacerbation
Asthma exacerbation was defined by the GINA guidelines as episodes of a progressive increase in shortness of breath, cough, wheezing, or chest tightness, or some combinations of these symptoms, accompanied by decreases in expiratory airflow and use of systemic corticosteroids (tablets, suspension, or injection), or an increase from a stable maintenance dose, for at least 3 days, and a hospitalization or emergency department visit because of asthma requiring systemic corticosteroids. Subjects were followed >2 years.
Animals and experimental protocol
All experimental animals used in this study were under a protocol approved by the Institutional Animal Care and Use Committee of Soonchunhyang University Bucheon Hospital. Female, 6 week-old BALB/c mice were sensitized and challenged with OVA as previously described.25 Airway responsiveness was measured, bronchoalveolar lavage fluid (BALF) was collected, and lung tissue was processed for protein, RNA, and hematoxylin and eosin (H&E) stain, and confocal imaging as previously described.25
Cell culture
Primary normal human bronchial epithelial (NHBE) cells (Lonza, Walkersville, MD, USA) were maintained (37℃, pH=7.4) in serum-free bronchial epithelial cell growth medium (BEGM, Lonza, Walkersville, MD, USA) supplemented with bovine pituitary extract, insulin, hydrocortisone, gentamicin/amphotericin, retinoic acid, transferrin, epinephrine, and human epithelial growth factor. NHBE cells were used before passage 7. Cells were placed in BEGM without supplements for 24 hour and then stimulated with 10 µg/mL house dust mite Dermatophagoides pteronyssinus peptidase 1 (Der p1) (Arthropods of Medical Importance Resource Bank, Institute of Tropical Medicine, Yonsei University, Seoul, Korea) with or without 10 µM dexamethasone (DEX) for 4, 8, or 24 hours. In separated tests, NHBE were transfected with small interfering RNA (siRNA) duplexes designed against claudin-4 or nonspecific siRNA control (Invitrogen, Carlsbad, CA, USA). NHBE cells cultured in 6-well plates were transfected with 100 nM siRNA or negative control using Lipofectamine 2000 (Invitrogen). After 24 hours, cells were treated with 10 µg/mL Der p1 and harvested for PCR analysis. Trans-epithelial electrical resistance measurements (TEER) was used as a measure of TJ formation in NHBE cells as previously described.25
Western blot analysis
Protein extracts of mouse lung tissue were collected as previously described.25 Protein was separated by SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked for 5% bovine serum albumin (BSA) in 0.1% Tween 20 in Tris-buffered saline (TBS) (21℃, 2 hours) and incubated with anti- claudin-4 (1:200, Abcam Inc., Cambridge, MA, USA) (4℃, overnight), followed by horseradish peroxidase (HRP)-conjugated secondary antibodies. Detection was performed using EzWestLumi plus (ATTO Corp, Tokyo, Japan). The relative abundance of protein was determined by quantitative densitometry, and the data were normalized to β-actin (Sigma-Aldrich, St. Louis, MO, USA).
Immunohistochemistry
Mouse lung sections were made as previously described,25 and then treated for non-specific binding with 1.5% goat serum and incubated with the anti-claudin-4 (1:100, Abcam). The next day, sections were incubated with avidin and biotinylated horseradish peroxidase macromolecular complex (Vector Laboratories, Burlingame, CA, USA). Color reaction was developed by staining with a liquid DAB + substrate kit (Golden Bridge International Inc., Mukilteo, WA, USA). After immnohistochemical staining, the slides were counterstaining with Herris's hematoxylin for 1 minute. Images were analyzed with the Image J program (National Institutes of Health, Bethesda, MD, USA), and stain density was quantified with an average of claudin-4 arbitrary density numbers from 6–8 fields.
Immunofluorescence imaging
Mouse lung sections were made as previously described.25 The sections were blocked for non-specific binding with 1.5% goat serum and incubated with the claudin-4 (1:400, Abcam Inc., Cambridge, MA, USA) +/- TJ protein 1 (TJP1 aka zonula occludens-1, ZO-1) (1:1,000, Santa Cruz Biotech, Santa Cruz, CA, USA), followed by Alexa Fluor 488-conjugated Donkey polyclonal anti-Rabbit IgG (1:1,000, Abcam Inc.) + PE-conjugated goat anti-mouse antibody (1:2,000, BD Bioscience). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (1:1,000, Invitrogen). Sections were observed using a confocal laser scanning microscope (LSM510 META), and images were generated using a Zeiss LSM image browser (Carl Zeiss Microsystems, Thornwood, NY, USA).
Quantitative real-time polymerase chain reaction (qRT-PCR) analysis
Total RNA was isolated using TRI REAGENT (Molecular Research Center, Cincinnati, OH, USA). For human cells and mouse lung RNA, cDNA was prepared from 3 µg RNA using oligo (dT), RNase out, and Superscript II reverse transcriptase (Invitrogen) (42℃, 50 minutes), followed by heating inactivation (70℃, 15 minutes). PCR was performed as previously described.25 The following thermal conditions were used: denaturation 94℃×5 minutes, followed by 30 cycles of 94℃×30 seconds, 60℃×30 seconds, and 72℃×30 seconds, and final extension at 72℃×7 minutes. Amplified PCR products were electrophoresed on 1% agarose gels, visualized using an ethidium bromide stain, and analyzed using Kodak EDAS 1D software. Alternatively, qRT-PCR was performed with the StepOne™ Real-Time PCR System (Applied Biosystems, CA, USA). The reactions were prepared with 20 µL of PCR mixture according to the manufacturer's protocol. The assay-on-demand gene expression products (Applied Biosystems, Inc.) were used to evaluate the mRNA expression levels of claudin-4, interleukin-4 (IL-4), IL-5, and IL-13. Target mRNA levels were normalized to PGK1 levels, and the ratios of normalized mRNA to untreated control sample were determined using the comparative Ct(2−ΔΔCt) method.
ELISA
Protein levels of IL-4, IL-5, and IL-13 or claudin-4 in mouse BALF or human plasma were measured by ELISA (R&D Systems, Mineapolis, MN, USA). To compare results from different plates, test sample ODs were adjusted relative to the positive and negative controls. The mean OD of duplicate wells was calculated. The index value of each tested serum was defined by the following formula: index=(OD of tested serum - OD of negative control)/(OD of the positive control - OD of the negative control)×100. Low detection limits were set at 2, 7, and 1.5 pg/mL or 0.066 ng/mL for IL-4, IL-5, IL-13, or claudin-4, respectively according to the manufacturer's recommendation.
Statistical analysis
Data are expressed as means±standard deviation (SD). For nonparametric data, the Mann-Whitney U test was used to assess differences between asthmatic and control groups, and followed by a post hoc test when appropriate (SPSS version 22; SPSS, Chicago, IL, USA). The comparison between stable and exacerbated asthma patients was made within subjects.
Correlations were evaluated by calculating Pearson or Spearman correlation coefficients. A Values of P<0.05 (two-sided) was deemed to indicate a statistical significance.
Go to :
RESULTS
Patients with asthma characteristics
Fifty asthmatic patients (mean age±SD, 54.9±14.1 years) and 25 control subjects (mean age±SD, 58.3±6.2 years) are shown in Table. Initial FEV1% pred, FVC% pred, and FEV1/FVC in patients with asthma significantly lower than in control subjects. Total IgE. atopy, and blood eosinophil proportion were significantly higher in patients with asthma than in control subjects. Body mass index was not different between asthmatic patients and control subjects. Duration of asthma was 6.63±3.60 years and the number of exacerbations per year during the follow-up was 3.38±3.24. FEV1% pred, FVC% pred, and FEV1/FVC were significantly lower in exacerbated asthmatics than in patients with stable asthma. Blood neutrophil proportion was significantly higher in exacerbated asthmatics than in patients with stable asthma.
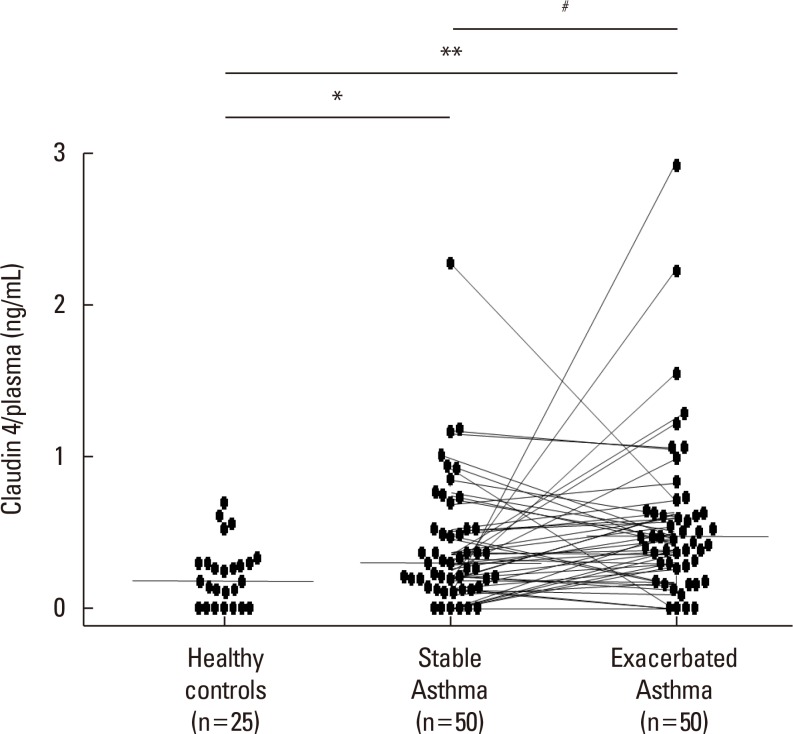
Alterations in claudin-4 in patients with asthma
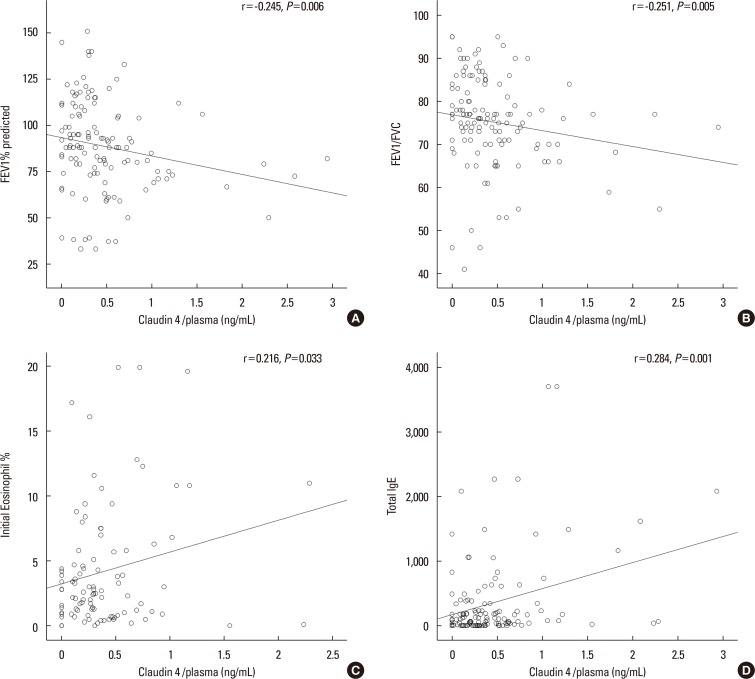
The mean plasma claudin-4 levels were 0.314±0.044 ng/mL in patients with bronchial asthma and 0.166±0.03 ng/mL in control subjects. The mean plasma claudin-4 levels were 0.451±0.061 ng/mL in exacerbated and 0.314±0.044 ng/mL in patients with stable asthma. Plasma claudin-4 levels were significantly higher in exacerbated patients than in patients with stable asthma (P<0.001, Fig. 1). The plasma claudin-4 levels were significantly higher in patients with stable asthma than in control subjects (P<0.001, Fig. 1). The plasma claudin-4 level was correlated with FEV1% pred (r=-0.245, P=0.006; Fig. 2A), FEV1/FVC (r=0.251, P=0.005; Fig. 2B), eosinophils (r=0.216, P=0.033; Fig. 2C), and total IgE (r=0.284, P=0.001; Fig. 2D).
OVA-induced inflammation, cytokines, and AHR in mice
The OVA-sensitized/challenged mice had increased AHR compared to control mice (Fig. 3A). The OVA-sensitized/challenged mice had increased inflammatory cells in BALF compared to control mice (Fig. 3B). IL-4 (36±3 pg/mL) and IL-5 (20±2 pg/mL) increased in BALF of the OVA-sensitized/challenged mice compared to the control mice (Fig. 3C).
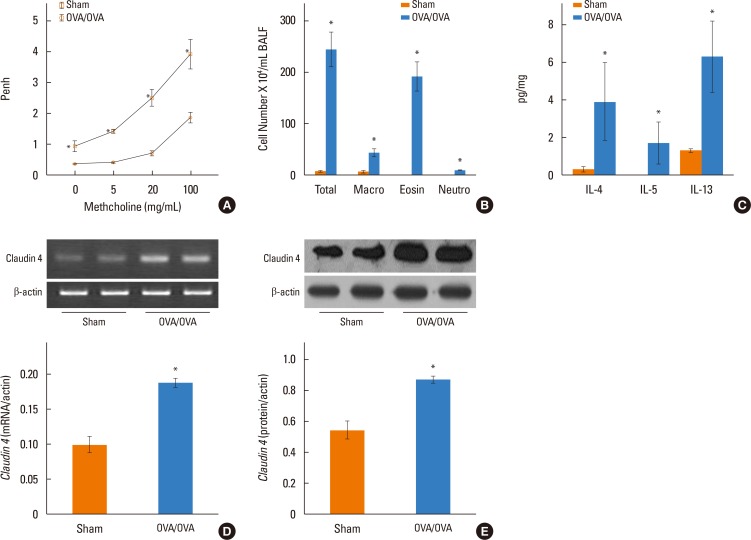 | Fig. 3Lung claudin-4 transcripts and protein levels in ovalbumin-sensitized and -challenged (OVA) mice. (A) Penh was measured following inhalation of increasing doses of methacholine, (B) Numbers of bronchoalveolar lavage fluid (BALF) cells, (C) Analysis of BALF cytokines, (D) Lung claudin-4 transcripts, (E) Protein levels. Densitometry was determined with 3 immunoblots and normalized to β-actin. Values (normalized to β-actin) means±SEM. *P<0.05 OVA vs sham.
|
OVA-induced inflammatory infiltrates and claudin-4 expression transcript/protein in mouse lung
Lung claudin-4 transcripts (Fig. 3D) and proteins (Fig. 3E) increased in the OVA-sensitized/challenged mice compared to saline-treated mice. On histologic examination, the OVA-sensitized/challenged mice had numerous focal regions with inflammatory cell infiltrates and peribronchial/intraluminal areas of exudation (Fig. 4A). The semi-quantitative value of the inflammatory index from H&E-stained images was increased in the OVA-sensitized/challenged mice. Increased claudin-4 immunohistochemical staining was noted in mononuclear inflammatory cells, endothelial cells, and epithelial cells from the OVA-sensitized/challenged mice (Fig. 4A).
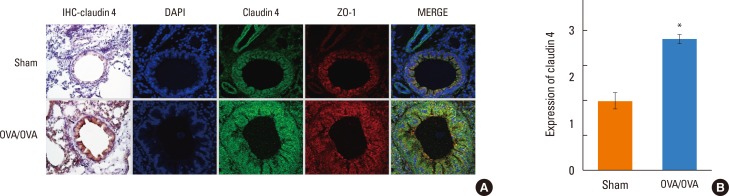 | Fig. 4(A) Immunohistochemistry and immunofluorescence staining of the lung sections of OVA/OVA and sham mice. Disrupted integrity of claudin-4 in the lung of the ovalbumin-sensitized and -challenged (OVA) mice. Tight junctions form a continuous ring that circumscribes individual cells is more evident but appear disrupted in the NHBE cells of OVA-sensitized/challenged mice. Disrupted integrity of claudin-4 in the lung. (B) Quantitation of claudin-4 was determined with 3 immunostains. *P<0.05 OVA/OVA vs sham.
|
The staining pattern of claudin-4 was altered in NHBE cells of the OVA-sensitized/challenged mice (Fig. 4). Normally, TJs form a continuous ring that circumscribes each cell. TJ rings became disrupted in the OVA-sensitized/challenged mice. Confocal image analysis showed significant increases in TJ breaks and the densities of claudin-4 staining (Fig. 4B), suggesting that disruption of the integrity of TJ proteins.
Der p1 increases claudin-4 transcripts and decreases TEER in NHBE
House dust mite Der p1, an aeroallergen with protease activity. To examine whether Der p1 could alter normal airway cells, NHBE cells were treated with 10 µg/mL Der p1 for 4, 8, or 24 hours and claudin-4 transcript levels were measured. Claudin-4 transcripts increased in NHBE cells following 10 µg/mL house dust mite Der p1 treatment for 8 hours (Fig. 5B). Der p1 increased TEER (Fig. 5C). Treatment of the cells with dexamethasone delayed the Der p1-induced TEER. When claudin-4 was decreased by siRNA transfection, the TEER was more decreased than scrambled siRNA control and the Der p1-induced increase in TEER was inhibited. At various time points, cytokine transcripts were increased in NHBE cells following Der p1 treatment (Fig. 6). Treatment of the cells with dexamethasone diminished this effect. The levels of IL-4, IL-5, and IL-13 transcripts were more increased in cells treated with Der p1 and siRNA directed at claudin-4 than in those treated with Der p1 alone. Treatment of the cells with dexamethasone inhibited this effect.
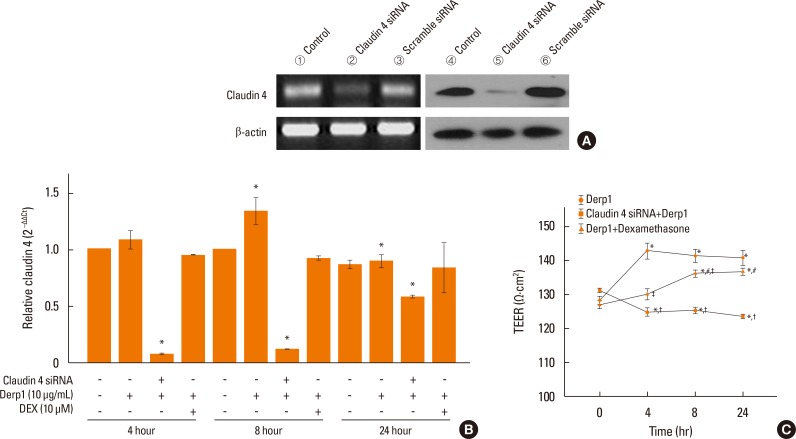 | Fig. 5Effects of claudin-4 knockdown on Der p1-induced inflammatory responses. (A) Representative image of siRNA directed claudin-4 in NHBE cells decreased (left panel) transcript determined by PCR (right panel) proteins determined by Western blot. (B) Claudin-4 transcripts increased in NHBE cells following 10 µg/mL Der p1 treatment for 8 hour. This response was inhibited by 10 µM dexamethasone (DEX) treatment. Small interference RNA directed at claudin-4 decreased claudin-4 mRNA. (C) Trans-epithelial electrical resistance (TEER) increased in NHBE cells following 10 µg/mL Der p1 treatment. This response was delayed in NHBE cells treated with 10 µM DEX treatment. siRNA directed at claudin-4 diminished the Der p1-induced increase in TEER. *P<0.05.
|
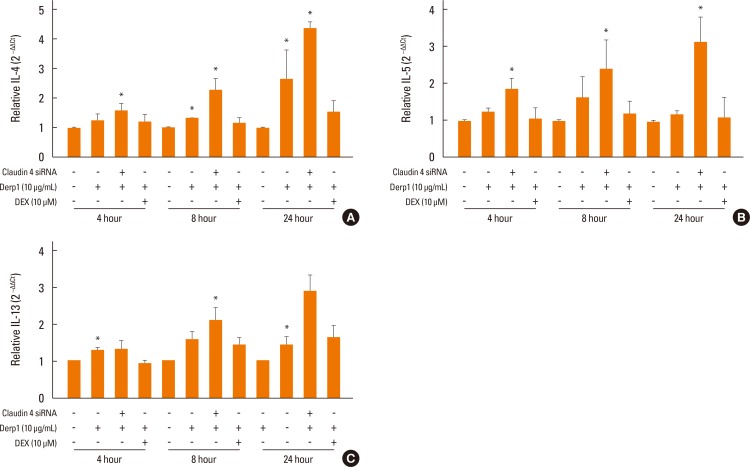 | Fig. 6Total RNAs extracted to measure mRNA expression levels of inflammatory cytokines (IL-4, 5, 13) by real-time PCR. (A) Interleukin 4 (IL-4), (B) IL-5 and (C) IL-13 transcript increased in NHBE cells following 10 µg/mL Der p1 treatment for 8 or 24 hours. This response was inhibited by 10 µM DEX treatment. The siRNA directed at claudin-4 increased the response to Derp1 stimulation. *P<0.05.
|
Go to :
DISCUSSION
Clinical manifestations of asthma, including episodic airway obstruction, airway hyperreactivity (AHR), and reduced lung function, are due to chronic airway inflammation and underlying structural changes in the airway wall.26 The bronchial epithelium forms a physical barrier of cell-cell junctional complexes and a chemical barrier of mucus which traps and removes inhaled particles by mucociliary clearance.27 As a gatekeeper to the environment, the bronchial epithelium plays a pivotal role in immune surveillance and appropriate activation of immune effector cells and antigen-presenting cells by controlling exposure to pathogen-associated molecular pattern molecules (PAMPs).27 It has been reported that altered lung endothelial CLDN5 expression was associated with airway inflammation in mice, and plasma CLDN5 levels were changed with asthma exacerbation, and asthma therapy can decrease plasma CLDN5 levels.25 Claudins are structural proteins of TJs, and different claudins are responsible for changes in the electrolyte and solute permeability across cell layers.28 Claudin-4 can function as a paracellular sodium barrier and is one of the 4 major claudins (claudins-3, -4, 7, and -18) expressed in lung epithelial cells.2128 The role of claudin-4 has been examined in acute lung injury,2123 pulmonary fibrosis,24 and lung cancer.29 However, its role in the pathogenesis of asthma is uncertain, and given that epithelial barrier functions can modulate immunity, we examined claudin-4 in this aspect.
To address this issue, 50 asthmatic subjects were recruited from the Genome Research Center for Allergy and Respiratory Diseases in Korea as previously described.25 Plasma claudin-4 levels were determined during periods when their asthma was controlled or during exacerbations. Initial FEV1 (%predicted), FVC (%predicted), and FEV1/FVC were significantly lower, and total IgE, atopy, and blood eosinophils were significantly greater in subjects with asthma than in those with control subjects. Among the subjects with asthma, FEV1, FVC, and FEV1/FVC were decreased and blood neutrophils were increased during exacerbations than during controlled state. Plasma claudin-4 levels were significantly greater in subjects with asthma than in those with controlled asthma. Plasma claudin-4 levels were increased further during asthma exacerbations. The plasma claudin-4 level was directly correlated with IgE and inversely correlated with FEV1 (%predicted), and FEV1/FVC. Those data suggest that claudin-4 may be useful as an epithelial biomarker for predicting clinical severity in asthmatic patients.
The OVA-sensitized/OVA-challenged mice had increased AHR, BALF inflammatory cells, and BALF cytokines as compared to the OVA-sensitized/saline-treated mice. Lung claudin-4 transcripts and proteins were increased in the OVA-sensitized/OVA-challenged mice compared to the OVA-sensitized/saline-treated mice. The OVA-sensitized/OVA-challenged mice had numerous focal regions with inflammatory cell infiltrates as well as peribronchial and intraluminal areas of exudation. The semiquantitative inflammatory index from H&E-stained images was increased in the OVA-sensitized/challenged mice. Increased claudin-4 immunohistochemical staining was noted in epithelial, mononuclear, and endothelial cells from the OVA-sensitized/OVA-challenged mice. TJs that form a continuous ring that circumscribes individual cells were disrupted in the OVA-sensitized/challenged mice. Confocal image analysis revealed significant increases in TJ disruption and claudin-4 staining. All animal model findings suggest that claudin-4, a TJ protein, is expressed epithelial cells, as well as inflammatory cells and disintegrated in asthma.
House dust mite allergens are important factors for the increasing prevalence of asthma. The lung epithelium forms a barrier that allergens must cross before they cause sensitization. The cysteine proteinase allergen Der p1 from fecal pellets of Dermatophagoides pteronyssinus causes disruption of intercellular TJs, which are the principal components of the epithelial paracellular permeability barrier.30 Claudin-4 transcripts are increased in NHBE cells following 10 µg/mL Der p1 treatment for 8 hours. At various time points, cytokine transcripts were increased in NHBE cells following Der p1 treatment. Treatment of cells with dexamethasone diminished this effect. The levels of IL-4, IL-5, and IL-13 transcripts were more increased in cells treated with Der p1 and siRNA directed at claudin-4 than in those treated with Der p1 alone. Treatment of cells with dexamethasone inhibited this effect. Der p1 increased in TEER. Treatment of cells with dexamethasone delayed the Der p1-induced TEER. When claudin-4 was decreased by siRNA transfection, TEER was decreased compared to scrambled siRNA control and the Der p1-indced increase in TEER was inhibited.
Inhaled corticosteroids are currently the most effective anti-inflammatory therapy for persistent asthma.26 Corticosteroid therapy can reduce airway inflammation, airway responsiveness, asthma symptoms, exacerbation frequency, and mortality. These effects are accompanied by the improvement of lung function and quality of life. Asthma exacerbation often results in excessive bronchospasm, mucus production, and bronchial edema. Corticosteroid therapy can diminish bronchial vasculature and edema in asthma. In NHBE cells, 10 µg/mL Der P1-induced TEER increase was delayed and cytokine transcripts were decreased by steroid treatment. A previous study has reported that higher doses of house dust mite extracts can cause transient decreases in TEER, which may depend on cell-cell interactions.30 Barrier function disruption is consistent with our in vivo findings in mice sensitized and challenged with OVA. Nonetheless, gene silencing of claudin-4 decreased baseline TEER and prevented the Der p1-induced increase in TEER, supporting a possible role of claudin-4 in the regulation of epithelial barrier function. Surprisingly, claudin-4 genesilencing leads to an increase in cytokine transcripts in NHBE following Der p1 treatment. This suggests a role of epithelial claudin-4 other than the protection of barrier functions, i.e. modulation of PAMP-induced cytokine generation and regulation of inflammatory cell functions.
The limitations of this study are the recruitment of clinical samples not matched for smoking status or sex, and the lack of data on claudin-4 from in vivo studies. Further studies are needed to confirm our results.
In summary, our data revealed that plasma claudin-4 levels were inversely correlated to lung function, suggesting that claudin-4 could be a marker for asthma inflammation and severity. Claudin-4 is altered in the epithelium from an OVA-sensitized/OVA-challenged asthma mouse model. A critical component of epithelial TJs and dysregulation of claudin-4 in the pulmonary epithelium can lead to cytokine release that in turn contributes to inflammatory cell activation and airway responsiveness. These effects can be repaired by steroid treatment. These findings thus raise the possibility that regulation of lung epithelial barrier proteins may constitute a therapeutic approach for asthma.
Go to :
 XML Download
XML Download