

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Respiratory viruses, predominantly rhinovirus (RV), are the most common cause of asthma exacerbation. Respiratory tract infections caused by respiratory viruses are associated with approximately 80% of asthma exacerbation episodes, in both adults and children123456 and RV is detected in approximately 60% of patients with asthma exacerbations.1 Patients with asthma are more susceptible to viral infections.7 Bronchial epithelial cells seem to be critical for the development of the innate immune response to virus infection,8 and some studies show a reduced generation of type I and III interferons (IFNs) in RV-infected bronchial epithelial cells (HBECs) from patients with asthma, reflecting an impaired antiviral response.9101112 Inadequate response to rhinovirus infection was also detected in other cells, such as alveolar macrophages,11 bronchoalveolar lavage cells,13 and blood mononuclear cells from asthma patients.101415 Similarly, reduced in vitro immune response to viral infections was observed in asthmatic children161718 and has been implicated in increased susceptibility to viral infections.1920 However, several other studies did not confirm reduced expression or release of IFNs in response to viral infection in HBECs from patients with asthma.2122232425 Several intracellular mechanisms are involved in the regulation of IFNs production by HBECs in response to virus infections,8 but activation of IFN-regulatory factors (IRFs) seems to play a central role. The IRF family includes IRF-1 to IRF-9, among which IRF-3 is expressed constitutively, while IRF-7 expression is induced by lipopolysaccharide and virus infection. Both IRF-3 and IRF-7 activate transcription of the genes for IFN-β, IFN-α1, and RANTES, while IRF-7 alone positively regulates genes for IFN-α4, IFN-α7, and IFN-α14.26 Structural cells, such as epithelial cells, produce IFN-β rather than IFN-α and IRF-3, which are essential for viral induction of IFN-β. IFN-α, is mainly produced by leukocytes and while IRF-7 can effectively induce genes for both IFN-α and IFN-β.2728 IRF-7 is mainly associated with IFN-α induction, as it is constitutively expressed in subsets of leukocytes, such as dendritic cells. The regulation of type III IFNs by viruses has been reported to also make use of IRF-3 and IRF-7.2930 IFN-γ-induced protein 10 (CXCL10, IP-10) and RANTES (CCL5) are released in great quantities by bronchial epithelial cells.31 It has been suggested that IP-10 release is specific to acute virus-induced asthma.3233 In contrast to rhinoviruses which have been extensively studied, immune response to parainfluenza viruses in HBEC from asthmatic patients has not been reported. Parainfluenza virus type 3 (PIV3) is a pleomorphic enveloped virus with nonsegmented negative-strand RNA genome (complementary to mRNA) which belongs to the Paramyxoviridae family and the Respirovirus genus. PIV3 is a common respiratory virus causing upper and lower respiratory tract illnesses, including common cold, laryngotracheobronchitis (i.e. croup), tracheobronchitis, bronchiolitis, and pneumonia, in both children and adults. Although these infections are rather mild in healthy individuals, they may lead to exacerbation in patients with asthma or COPD.34
In order to better understand determinants of variability in immune responses to respiratory virus infections of airway epithelial cells in patients with asthma, we evaluated the response to PIV3 and RV1B infection in primary bronchial epithelial cells cultured from the phenotypically heterogeneous group of asthmatics and healthy controls. These studies are necessary to ascertain the relative efficacy of inhaled IFN therapy for asthma35 and the role of atopy in influencing anti-viral immunity to respiratory viruses.
MATERIALS AND METHODS
Study subjects
Ten patients (6 atopic and 4 non-atopic) with bronchial asthma diagnosed according to the GINA criteria were recruited from the the Department of Pulmonology and Allergology. All patients suffered from persistent asthma and were treated with inhaled corticosteroids, which were withdrawn 24 hours before bronchial sampling. Asthma severity was assessed according to the GINA guideline36 and asthma control with the Asthma Control Test (ACT).37 Atopy was defined as the presence of at least 1 positive skin response (wheal diameter >3 mm) to skin-prick tests (SPTs) performed with the panel of 14 common inhalant allergens. Patient characteristics are presented in Table 1. Nine healthy volunteers aged 31.78±14.62 with no previous history of any lung disease and with negative SPTs to a panel of inhaled allergens served as healthy controls. The study was approved by the local Bioethics Committee, and all study subjects provided informed consent.
Bronchial epithelial cell tissue culture
HBECs were grown from brushings of the bronchial tree during bronchoscopy. HBECs were cultured as monolayer in hormone-supplemented, serum-free bronchial epithelial growth medium (Lonza Inc., Allendale, NJ, USA). Cells were confirmed as epithelial by cytokeratin 19 staining and flow cytometry. Cells were split when confluent and seeded for experimentation at passage 2. HBECs were infected with RV1B or PIV3 (multiplicity of infection 0.1) for 1 hour. Virus was then removed, and cells were washed 3 times and replaced by fresh medium. After that, cells were harvested at 8, 24, and 48 hours. As a negative control, cells were treated with medium alone.
RNA isolation and quantitative PCR
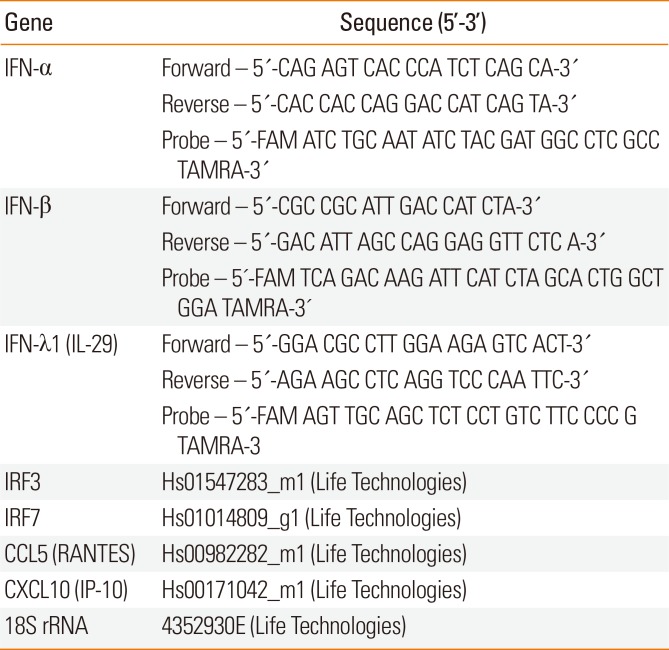
Total RNA was extracted from cells with the RNeasy MiniKit (Qiagen, Germantown, MD, USA). Then, cDNA was transcribed using the RevertAid H Minus First Strand cDNA Synthesis Kit (ThermoFisher Scientific, Waltham, MA, USA), followed by quantification of target mRNA using the TaqMan Universal PCR Master Mix (ThermoFisher Scientific). Expression of RV1B mRNA was quantified using the PrimerDesign Genesis Kit for Human RV1B, and expression of PIV3 mRNA was quantified using the PrimerDesign Genesis Kit for Human HPIV3 (Genesig Ltd, Lafayette, PA, USA). Relative expression of the following genes was quantified by the comparative CT (ΔΔCT) method using 18S rRNA as a reference gene: IFN-α, IFN-β, IFN-λ1 (IL-29), RANTES (CCL5), IP-10 (CXCL10), and IRF-3/IRF-7. Reactions were analyzed with the StepOnePlus Real-Time PCR System (ThermoFisher Scientific) at 50℃ for 2 minutes, 95℃ for 10 minutes, and 45 cycles of 95℃ for 15 seconds and 60℃ for 15 seconds. Each gene was normalized to 18S rRNA. Real-time RT-PCR was carried out using specific primers and probes for each gene (Table 2).
Measurement of IFN and chemokine protein
IFN-β (PBL Assay Science) and IFN-λ1 (eBioscience, San Diego, CA, USA) were measured by ELISA according to the manufacturer's instructions. The sensitivities of these assays were 50 and 15.6 pg/mL, respectively. The concentrations of IFN-α, IP-10, and RANTES in the culture supernatants were measured using BD Cytometric Bead Array (CBA) (Becton, Dickinson Co., Franklin Lakes, NJ, USA). The sensitivity of this assay was 10 pg/mL each.
Statistical analysis
All statistical analyses were performed with Statistica 10 (AXAP301E504323AR-B). Medians (interquartile ranges) were used instead of means (SDs) if data were non-normally distributed. Normality was tested with the Kolmogorov-Smirnov test with Lilliefors correction and the Shapiro-Wilk test. Student t test, the Mann-Whitney test, or the Kruskal-Wallis test with Dunn's multiple corrections test were used to determine differences in antiviral responses between the groups as appropriate. Correlations were analyzed using the Spearman rank test. A P value of <0.05 was considered statistically significant.
RESULTS
PIV3 and RV1B replication in HBECs
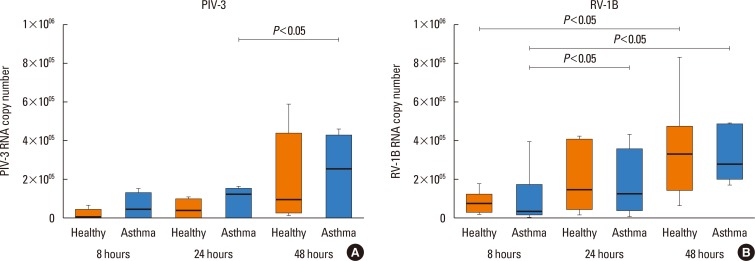
PIV3 mRNA was detected in cell lysates of HBECs from both asthmatic patients and healthy controls at all time points (8, 24, and 48 hours) after infection (but not in control, non-infected cells) (Fig. 1A and B). A significant increase in PIV3 gene copy number at 48 hours compared to 24 hours was observed only in asthmatic patients (P<0.05). RV1B mRNA was detected in HBECs from both groups at all time points after infection (but not in control, non-infected cells), and the RV1B gene copy number was increased in subjects of both groups reaching significance at 48 hours compared to 8 hours (P<0.05). The Replication rate of either PIV3 or RV1B in HBECs was not significantly different between subjects with and without asthma at any time point. Since virus replication was assessed only in 5 asthmatics, we could not compare atopic vs non-atopic asthmatics.
IFN generation in response to virus infection in HBECs from patients with asthma and in healthy controls
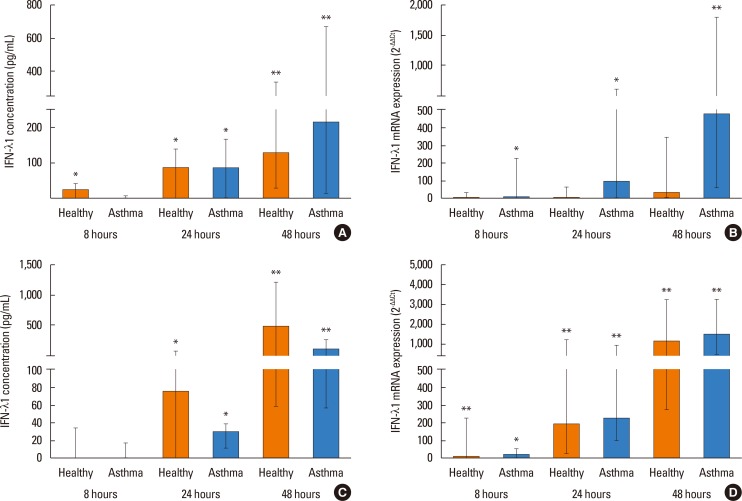
PIV3 and RV1B infection induced a significant IFN-λ1 protein increase in cell supernatants from asthmatics as well as healthy controls (Fig. 2A and C). There was no significant difference between healthy controls and asthmatics in the amount of IFN-λ1 protein released, at any time point. IFN-λ1 mRNA expression was significantly induced by PIV3 infection in patients with asthma at all time points compared to medium control (Fig. 2B). PIV3 induced IFN-λ1 mRNA expression in asthmatics, but not significantly in healthy controls. Infection with RV1B induced IFN-λ1 mRNA expression in both subjects with asthma and healthy individuals (Fig. 2D). There was no difference in the magnitude of virus-induced mRNA expression between asthmatics and healthy controls for either virus.
IFN-α and IFN-β proteins were not detectable in cell supernatants after either PIV3 or RV1B infection. Both viruses induced IFN-α mRNA expression at 48 post infection in subjects with asthma, but not in healthy controls. Following PIV3 infection IFN-β mRNA expression was significantly induced only in asthmatic patients at 48 hours, but RV1B induced IFN-β mRNA in both study groups at 24 hours (P<0.05) and 48 hours (P<0.01). There was no significant difference between subjects with and without asthma in IFN-α or IFN-β mRNA expression after either PIV3 or RV1B infection.
Expression of IRFs
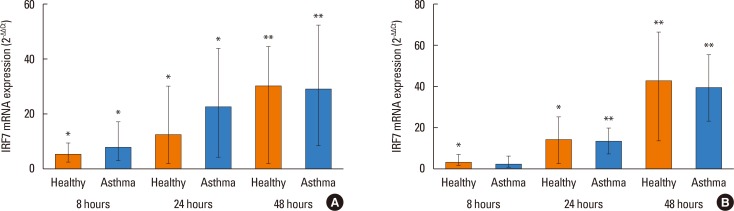
We next assessed the expression of IRF-3 and IRF-7 in virus-infected bronchial epithelial cells. In parallel with IFN-λ1 release, there was significant induction of IRF-7 mRNA by both PIV3 and RV1B at all time points without difference between asthmatics and healthy controls (Fig. 3A and B). We did not observe IRF-3 expression after infection with either of the viruses (data not shown).
There was a strong positive correlation between IRF-7 mRNA of and IFN-λ1 mRNA expression in both healthy controls and asthmatic following PIV3 infection, while after RV1B infection such correlation was observed only in healthy controls. Interestingly after RV1B infection, a correlation between IRF-7 and IFN-β gene expression was observed in asthmatics at 24 hours (r=0.63, P<0.05) and 48 hours (r=0.73, P<0.05).
IP-10 and RANTES generation in response to virus infection in HBECs from patients with asthma and in healthy controls
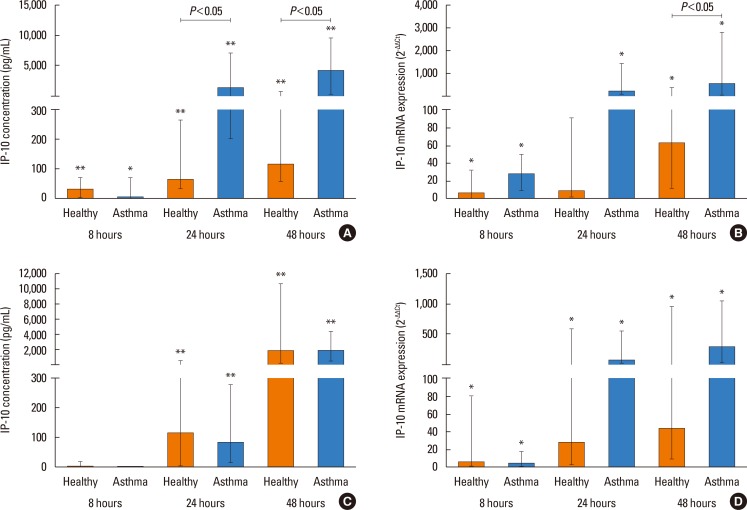
PIV3 induced significant release of IP-10 protein into cell supernatants of HBECs in both healthy controls and asthmatics; however, HBECs from asthmatics produced significantly more IP-10 protein at 24 hours (median 1,296.65 pg/mL [256.17–5,740.30]) and 48 hours (median 4,119.34 pg/mL [421.82–9,366.14]) compared to HBECs from healthy controls (64.64 pg/mL [45.91–91.12], 111.24 pg/mL [64.57–514.52]) at 24 and 48 hours, respectively (P<0.05) (Fig. 4A). In parallel, IP-10 mRNA expression was significantly higher (median 8.26-fold) in asthmatics (572.51 [216.77–1,078.64]) compared to HBECs from healthy controls (61.83 [18.31–302.85], P<0.05) at 48 hours (Fig. 4B). RV1B infection induced IP-10 release at 24 and 48 hours (P<0.01) in both groups and a trend toward higher expression of IP-10 mRNA was observed in asthmatics (Fig. 4C and D). Both PIV3 and RV1B infection induced significant (P<0.01) RANTES release, which was similar in epithelial cells from both groups (data not shown).
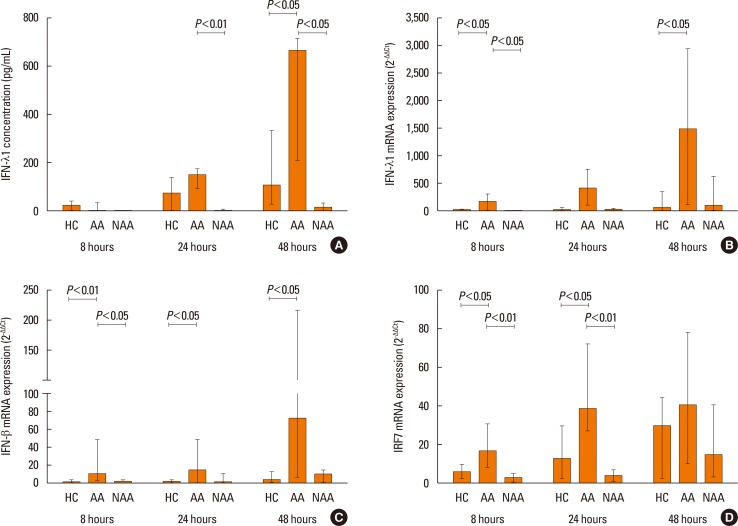
IFN generation in response to virus infection in HBECs from atopic and non-atopic asthmatics
In patients with non-atopic asthma (n=4), IFN-λ1 protein production in response to PIV3 was supressed at 24 and 48 hours, which was significantly lower (P<0.05) compared to subjects with atopic asthma (n=6) (Fig. 5A). The IFN-λ1 protein level in response to PIV3 in atopic asthmatics was significantly higher at 48 hours compared to non-atopic asthmatics (5.60-times) and healthy controls (29.09-fold) (Fig. 5A). IFN-λ1 mRNA expression was significantly higher in atopic asthmatics at 8 hours compared to non-atopic asthmatics and healthy controls and at 48 hours post infection compared to healthy controls (Fig. 5B). In atopic asthmatics (but not in non-atopic), IFN-β mRNA induction by PIV3 was significantly higher compared to healthy controls at all time points (Fig. 5C). No difference in IFN-α mRNA expression of HBECs between patients with atopic or non-atopic asthma was observed.
IRF-7 mRNA induction was significantly lower in HBECs from non-asthmatics compared to those from atopic asthmatics at 8 and 24 hours (P<0.01) post PIV3 infection; however, only in atopic asthmatics PIV3 induced IRF-7 mRNA expression was higher compared to healthy controls at 8 and 24 hours (P<0.05) after infection (Fig. 5D).
Following RV1B infection, atopic asthmatics had significantly higher IFN-β mRNA expression at 24 hours (P<0.05) and 48 hours (P<0.01) compared to non-atopic asthmatics; however, no significant differences between groups were observed with respect to IFN-λ1 protein generation or IFN-λ1, IFN-α, and IRF-7 mRNA expressions (data not shown).
IP-10 and RANTES generation in response to virus infection in HBECs from atopic and non-atopic asthmatics
Following PIV3 infection, RANTES protein levels (but not mRNA expressions) in non-atopic asthmatic patients were significantly lower at 8 hours (median 0.40 [0.34–0.49] pg/mL) compared to atopic asthmatics (median 8.87 [5.50–16.87] pg/mL, P<0.01) or healthy subjects (median 10.29 [3.65–19.22] pg/mL, P<0.01). However, there was no significant difference between .atopic and non-atopic asthmatic patients in IP-10 protein synthesis or IP-10 mRNA expression at any time point (data not shown). Following RV1B infection, there was no difference in IP-10 or RANTES (protein or mRNA) expression between atopic and non-atopic asthmatics (data not shown).
Immune response to virus infection in HBECs from patients with severe and non-severe asthma
Patients with severe asthma, defined according to the GINA criteria, had significantly lower ACT (11.75±2.75) compared to non-severe asthmatics (20.83±1.62, P<0.01); however, there was no significant difference either in PIV3 or RV1B induced IFN-λ1 protein and mRNA or in IFN-α and IFN-β mRNA expressions in HBECs between asthmatics with severe (n=4) and non-severe asthma (n=6). Expression of IRF-7 was similar in both groups. There was no difference in IP-10 or RANTES protein level or gene expression after infection with either virus between patients with severe or non-severe asthma.
DISCUSSION
Over the last decade, deficient innate immune response (including IFN production) to virus infection in HBECs from patients with asthma has been observed since the original report by Wark et al.,9 but it has not been confirmed in all the studies. Most reported studies employed a single virus (usually one of the RVs) and these studies differed in the experimental technique, including culture method, virus load, and clinical characteristics of the subjects, which may be responsible for the conflicting results. In this study, we assessed in parallel innate immune response to 2 respiratory viruses (PIV3 and RV1B) in primary HBECs cultured from a phenotypically heterogeneous, but well characterized group of asthma patients and healthy controls.
Initially, we demonstrated that the viral replication rates for both PIV3 and RV1B in HBECs from asthmatic patients and healthy controls were not different. Our observation is in line with other in vitro studies.212224 However, other studies showed increased RV replication in asthmatic HBECs ex vivo9111617 and in vivo using experimental virus challenge.3839 Similarly, Patel et al.25 documented higher levels of influenza A virus protein, but not RSV in HBECs from asthmatics compared to healthy controls.
Furthermore, we did not find any quantitative difference between asthmatic patients and healthy controls in the magnitude of IFN response to either virus. Consequently, IRF-7, an inducible interferon-stimulated gene in bronchial epithelial cells, was increased in parallel with the infection and was augmented to a similar extent in both groups. Previous studies reported type I or III IFN induction in primary HBECs of asthmatic patients in response to RV infection lower911121617 or the same21222425 compared to healthy controls. Impaired immune responses were also observed in HBECs stimulated with viral dsRNA.4041 Although deficient IFN type I and III response has been identified in PBMCs from pregnant female asthmatics infected with RV-43 or RV1B, no difference was observed between non-pregnant female asthmatics and non-pregnant healthy females.42 In a recent study, Sykes et al.24 showed that in relatively mild and well-controlled asthmatics, IFN production in HBECs in response to RV16 was not impaired. Contradictory results of studies reporting either virus replication or IFN responses to virus infections in patients with asthma may reflect the phenotypic heterogeneity of the disease, reflected also by variability in the innate immune response. Our asthmatic patients were quite heterogeneous to some clinical traits, such as presence of atopic sensitization or severity of the disease. Thus, in order to gain some insight into the potential role of host factors on ability of HBECs to respond to viral infections, asthmatic patients were stratified according to pathophysiological and clinical phenotypes. Compared to atopic patients, HBECs from subjects with non-atopic asthma had significantly diminished overall immune response to PIV3. At 48 hours post infection, IFN-λ1 protein was barely detectable in supernatants of HBECs from non-atopic asthmatics, while in atopic asthmatics its level was very high (median 660.04 pg/mL) and significantly higher compared to healthy controls. Differences in IFN-λ1 protein release between non-atopic and atopic asthmatics were paralleled by a similar pattern of IFN-λ1 mRNA expression. Although IFN-β protein was not detectable, there was a transient increase in IFN-β mRNA expression in atopic asthmatics at 8 hours post infection with PIV3 compared to non-atopic asthmatics. Impairment in IFN expression in non-atopic asthmatics was paralleled by a decrease in expression of IRF-7 with significant differences from atopics at 8 and 24 hours after PIV3 infection. In contrast, chemokine (IP-10 and RANTES) release did not differ between atopic and non-atopic asthmatics.
In response to RV1B, no difference in IFN, IRF, or chemokine expression between atopic and non-atopic asthma patients were observed, with the exception of IFN-β mRNA, which was significantly lower in non-atopic asthmatics. The lack of difference between atopic and non-atopic asthmatics in IFN-λ1 release and expression in response to RV1B may result from different pathophysiological mechanisms of epithelial immune response to different viruses.4344 Alternatively, it is possible that the difference was not detected due to significantly weaker overall immune response of epithelial cells to RV1B compared to PIV3; the experiments should be repeated with higher RV MOIs.
Our results suggest that non-atopic status of asthma patients which is distinguished by lack of allergic sensitisation may be associated with impaired IFN (but not other chemokines like RANTES) response to virus infection. In contrast, in atopic asthmatics IFN-λ1 and IFN-β response to PIV3 infection was either comparable or even more intense compared to healthy controls. Non-atopic asthma is not a well-characterized phenotype and is rather diagnosed by exclusion of atopic sensitization. The inflammatory process in the airways seems to be very similar between allergic and non-allergic asthma based on the expression of inflammatory mediators, including Th2 cytokines and eosinophilotactic chemokines, but prevailing triggering factors seem to be different.45 On the other hand, a significant heterogeneity of asthma may involve different inflammatory phenotypes, some of which may differ in terms of innate immune activation during viral infection.46 Our observations documenting impaired IFN responses to viral infection in a subpopulation of asthma patients are in line with well-established non-atopic asthma phenotype, which includes respiratory infections as dominant exacerbation-triggering factors.47 A recent systematic review documented association of non-atopic asthma/wheeze with lower respiratory tract infections in children and adolescents.48 Interestingly, non-atopic asthmatics are more likely to have respiratory pathogens detected in nasopharyngeal swabs during asthma exacerbation.49 Thus, further studies are necessary to establish the immune response in atopic asthmatics compared to non-atopic asthmatics.
However, a significant limitation of this analysis is related to relatively small sample size, with 6 participants in atopic and 4 in non-atopic asthmatic and further heterogeneity within subgroups. The differences were not related to asthma control or severity, since asthma control test and overall severity according to GINA criteria or maintenance dose of inhaled corticosteroids were similar in atopic and not atopic asthma patients. However, non-atopic asthmatics were on average older and had significantly lower respiratory function parameters compared to atopic asthmatics, which may have affected immune response to viral infection. These results cannot be easily referred to other studies assessing antiviral response in HBECs, since in earlier studies either only atopic asthmatics were recruited91117212224 or patients' atopic status was not reported.12 The antiviral response to respiratory virus in patients with atopic and non-atopic asthma was reported by Baraldo et al.16 who demonstrated impaired expression of IFN-β gene after infection with RV16 in children with asthma compared to controls. At the same time, they did not find any differences in the antiviral response between children with atopic and non-atopic asthma.
Although generation of IFNs and RANTES in response to infection with PIV3 or RV1B was similar in the whole group of asthmatics and healthy controls, significant differences were observed with respect to IP-10 production. Upon PIV3 infection, patients with asthma expressed significantly more IP-10 both at protein and mRNA level than healthy controls. A similar trend toward higher expression of IP-10 was also observed after RV1B infection; however, the difference was not statistically significant. IFN-γ-IP-10 (CXCL10) belongs to the CXC chemokine subfamily that binds to common CXCR3 receptor and is produced by various cell types, including airway epithelial cells in response to viral infection.50 Previously, significantly increased serum IP-10 levels were observed in subjects with acute virus-induced asthma compared to non-virus-induced acute or controlled asthma,3233 and RV-infected human airway epithelial cells produce IP-10 in vitro and in vivo.31 However, Wark et al.32 did not observe significant differences in IP-10 serum levels in asthmatic patients compared to healthy volunteers. Similarly, no differences in IP-10 release by nasal epithelial cells and tracheal epithelial cells from children with wheeze/atopy in response to RSV or hMPV infection were observed.56 IP-10 is thought to play an important role in the Th1-mediated immune response, but it has been implicated also in exacerbation of Th2-mediated airway inflammation typical for bronchial asthma.5152 Th2 cytokines increase release of IP-10 and other inflammatory cytokines in the presence of RV infection.53 Since there is evidence that IP-10 is generated downstream to IFNs and that IFN-λ can also induce IP-10 production, elevated IP-10 generation (but not IFN production) in asthma patients might be related to other mechanisms and warrant further investigation.
Comparing immune/inflammatory response between different respiratory viruses is controversial, since even different RV strains can induce different patterns of cytokines and chemokines.5455 Thus, it is important to assess innate immune response to various respiratory pathogens in asthmatics. Previous studies reported innate response to several respiratory viruses e.g. RV, RSV,2526 hMPV,56 and influenza A2557 in airway epithelial cells from asthmatic patients, but this is the first report documenting that PIV3 efficiently replicates in bronchial epithelium from asthmatics. The role of type III INFs in the immune response to PIV3 has not been studied in patients with asthma. In this study, we demonstrated that IFN-λ is the major IFN subtype induced in HBECs by PIV3 infection, similar to other respiratory viruses.102224 As in studies with other respiratory viruses,1024 we were able to detect IFN-β and IFN-α only at the mRNA level. However, in our model PIV3 and RV1B induced type I INFs only in patients with asthma (data not shown).
The increase in IFN production by PIV3 and RV1B was associated with parallel induction of IRF-7 mRNA which was observed as early as 8 hours post infection and sustained up to 48 hours. IRF-7 mRNA expression following PIV3 infection highly correlated with IFN-λ1 expression in both healthy controls and asthmatics, while after RV1B infection a significant correlation was observed only in healthy controls. On the other hand, IRF-7 expression correlated with IFN-β expression in response to both viruses in asthmatics patients and after RV1B only in asthmatics. As expected, expression of IRF-3 mRNA in HBECs infected with respiratory viruses was not changed, since activation of IRF-3 is associated with increased phosphorylation rather than changes in expression.
In summary, this is the first report documenting that PIV3 efficiently replicates in primary HBECs from patients with asthma and that IFN-λ1 release and expression in epithelial cells, which are associated with IRF-7 expression, are pivotal to the antiviral response. However, the most intriguing observation is the modulatory effect of atopy on the impaired generation of INFs in the airway epithelium of asthmatic patients in response to virus infection. Further studies are warranted to confirm if such an effect can be observed with other viruses and to elucidate the significance of this phenomenon in the pathogenesis of respiratory virus infections in asthmatic patients.
 XML Download
XML Download