

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Bronchial asthma is a common chronic inflammatory disorder of the airways that is increasingly recognized as a heterogeneous clinical syndrome. Through a vast amount of research, current knowledge on the pathogenesis of asthma has shifted from a single disease due to airway obstruction to a complex disorder consisting of diverse phenotypes.1 The heterogeneous nature of bronchial asthma necessitates advancement in treatment, particularly for severe asthmatic patients refractory to conventional therapies, including inhaled or systemic corticosteroids (CS) and bronchodilators. Currently, most of the asthma treatment guidelines are based on the strategy which mainly targets T helper type 2 (TH2) cells-dominant airway inflammation and these are efficacious in many patients. However, 5%–10% asthmatic patients remain poorly managed by these modalities and account for about 50% of the health care expenditure for asthma care.2 Notably, severe asthma is also heterogeneous in nature, in which different cellular components and pathogenetic mechanisms contribute to the disease, requiring different therapeutic approaches.3 Therefore, defining etiologies and underlying pathobiologic mechanisms (i.e. endotypes) in various types of severe asthma is a major field of respiratory research.45
Respiratory fungal exposure is constant in humans, and fungal spores constitute the largest proportion of aerobiological particles in usual air environment.6 Thus, the impact of respiratory fungal exposure on clinical courses of bronchial asthma has been widely reported in the literature for a long time.78 In particular, fungal exposure has been reported to be associated with the development of asthma.9 Furthermore, exposure to environmental fungal spores also leads to the exacerbation of allergic symptoms and asthma.1011 These results suggest the crucial role of fungi in allergic lung inflammation, especially in the more severe phenotype of the disease. Importantly, this idea is further supported by the fact that fungi can colonize, actively germinate, and infect the human respiratory tract, thereby potently sensitizing and inducing the host immune response.712 Moreover, they can produce a wide array of enzymes and toxins closely implicated in allergic inflammation.13 In this context, epidemiologic studies have also shown that fungal sensitivity is a possible precipitating factor for life-threatening asthma.1415 Based on this knowledge, severe asthma with fungal sensitization (SAFS) has been proposed to be investigated as a particular phenotype of severe asthma with therapeutic implications in clinical trials.71617 However, treatment with antifungal agents in SAFS patients failed to show a consistent beneficial effect on the clinical course of the disease in 2 randomized clinical trials.1617 In fact, little is known about the exact role of fungi in severe allergic lung inflammation.
Recently, increasing evidence suggests that cellular stresses due to impaired biological processes can be integrated into inflammatory response, thereby being implicated in various human inflammatory disorders. Endoplasmic reticulum (ER) is the largest intracellular organelle which manipulates crucial soluble and membrane proteins, and it is abundantly equipped by numerous enzymes that facilitate proper folding of the client proteins.18 In this respect, stress response originates from the ER related to the accumulation of unfolded and/or misfolded proteins in the ER lumen (i.e. ER stress),19 has been increasingly reported to be involved in the pathogenesis of diverse inflammatory disorders.20 Furthermore, involvement of ER stress in the pathogenesis of numerous pulmonary disorders, including bronchial asthma, chronic obstructive pulmonary disease (COPD), fibrotic lung disorders, and acute lung injury (ALI)/acute respiratory distress syndrome (ARDS), has been widely investigated.2122232425
In this review, we focus on the emerging role of ER and associated cellular immune/inflammatory molecular platforms in the lung, particularly in the pathogenesis of fungal allergy and SAFS. Additionally, we present our recent data on these issues, which further highlight the therapeutic potential of ER stress in fungi-induced severe allergic inflammatory disorders in the lung.
SAFS and other fungal sensitization/allergy-associated clinical conditions
Fungal sensitization and allergy development refer to an exaggerated immune response typically against non-pathogenic fungi. Fungal sensitization and allergy are further distinguished by the presence of immune-mediated tissue damage because sensitization itself does not always cause inflammatory response.26 In contrast, the term fungal infection can be applied when there is evidence of tissue dysfunction associated with the growth and invasion of pathogenic fungi in the host. Whereas viable fungi are implicated in both allergy and infection, causative agents for allergy do not need to be viable to induce an allergic response (e.g. structural components and byproducts). There are several important disease entities representing the severe end of fungal sensitization/allergy development-associated conditions.26 These include allergic bronchopulmonary aspergillosis/mycosis (ABPA/ABPM) and SAFS.
ABPA is a complex hypersensitivity reaction that often occurs in patients with asthma or cystic fibrosis when bronchi become colonized by Aspergillus species, mostly Aspergillus fumigatus.27 In general, the diagnosis of ABPA is a composite of clinical, radiological, and immunologic features (Table). In the later courses of ABPA, repeated episodes of bronchial obstruction, inflammation, and mucoid impaction can lead to irreversible structural changes. Many patients with ABPA respond well to treatment with systemic CS, whereas some patients are poorly controlled and can be complicated by progression to bronchiectasis and pulmonary fibrosis. Similarly, fungi other than Aspergillus species, such as Candida albicans, can induce an ABPA-like disease process, which refers to ABPM.
Whereas ABPA was first reported in 1952, the definition of SAFS (Table) was introduced in 20067 and has been used in clinical trial settings to demonstrate the possible role of antifungal therapy in treating a particular phenotype of severe asthma associated with fungi.1617 In fact, ABPA/ABPM is an extreme spectrum of allergic inflammatory response against fungi, thus most patients sensitized to fungi without convincing evidence of lung damage had not been incorporated into a specific disease entity.26 Therefore, researchers have proposed SAFS that can be defined as patients having both severe asthma and evidence for fungal sensitization (i.e. positive skin prick test, positive fungal specific IgE in blood) without satisfying the criteria of ABPA.7 Thereafter, several clinical studies suggested the role of antifungal agents in the treatment of the SAFS patient group.1628
Meanwhile, most of the current diagnostic criteria for ABPA/ABPM have been in practice since the 1970s and do not precisely reflect the natural history of the disease, especially in its early phase.26 Consequently, these diagnostic criteria of ABPA/ABPM may not be effective for the prevention of permanent lung damage, such as bronchiectasis. Additionally, the definition of SAFS does not represent the direct causality of fungal sensitization in inducing severe asthma, even though it is convenient for patient inclusion in clinical trials. In this respect, SAFS was not proposed as an asthma endotype having a distinct pathobiological mechanism.4 Conflicting results from previous clinical trials, in which efficacies of antifungal agents in the treatment of SAFS were investigated,1617 may be partly due to the limitation in SAFS definition and unstandardized testing tools demonstrating fungal allergy. More precise definitions that represent the diverse spectrum of fungal allergy-associated clinical conditions are needed in the near future.
Interactions between fungi and host
Research on the mechanisms of fungi-associated disorders, including fungal allergy and infection, has revealed intricate molecular networks between the fungi and host. In particular, they mainly focused on what kind of fungi-derived molecules can be effectively recognized by the host immune system through what kind of innate receptors and how these receptor-ligand interactions can be interpreted and linked to appropriate host inflammatory responses.
Fungi contain various antigens in their cell wall. Among various components, polysaccharides, including α- and β-glucans, galactomannan, and chitin, are major constituents of fungal cell wall.29 In addition, fungi can produce large amounts of secreted enzymes, including proteases and glycosidases, which can directly damage host tissues.26 Both fungal cell wall and secreted components can induce protective host immune responses against fungi. At the same time, they also act as major fungal allergens in the lung.
Initial recognition of fungi
The complex interaction between the host and fungi begins with recognition of the fungal components, called pathogen-associated molecular patterns (PAMPs), through several evolutionarily conserved pattern recognition receptors (PRRs) of the innate immunity.30 The C-type lectin receptor (CLR), Toll-like receptor (TLR), and NOD-like receptor (NLR) families of PRRs are known to be involved in anti-fungal host immunity.
CLRs
Since about 60% of the dry weight of the fungal cell wall is β-glucans, glucan concentration in dust can be used as a surrogate marker for estimating fungal exposure.831 CLRs consist of a transmembrane receptor family having a carbohydrate-binding domain, thereby recognizing carbohydrates on the fungi.30 Among them, dectin-1 and dectin-2 expressed on macrophages, neutrophils, and dendritic cells (DCs) sense β-glucans of fungi and generate anti-fungal effector responses with phagocytosis, oxidative burst, and production of inflammatory mediators, including interleukin (IL)-6, IL-8, and tumor necrosis factor (TNF)-α.8 In CLR signaling, activation of downstream Syk tyrosine kinase is known to be integral in the activation of mitogen-activated protein (MAP) kinases and important transcription factors, including nuclear factor (NF)-κB through CARD9.29 The clinical importance of the CLR-associated pathway is further emphasized by the fact that mutations in dectin-1 and CARD9 lead to chronic fungal infection.3233 Moreover, considering that aerosol exposure of β-glucan resulted in pulmonary eosinophilia in an animal model34 and that the levels of β-glucan were positively associated with peak expiratory flow variability in children with asthma,35 the CLR pathway may play an important role in the pathogenesis of fungal allergic lung inflammation.
TLRs
TLRs are broadly expressed on nonprofessional innate cells, such as epithelial cells and fibroblasts, as well as innate immune cells. A wide range of fungal PAMPs (e.g. phospholipomannan, glucuronoxylomannan, O- and N-linked mannans, DNA, and RNA) are recognized by numerous TLRs.36 Their engagements with cognate TLRs activate intracellular pathways via association with the myeloid differentiation primary response gene 88 (Myd88) (except for the TLR3 pathway), a key downstream adaptor for most TLRs and IL-1 receptors,30 and intersect with cellular adaptor proteins and important transcription factors, such as NF-κB.36 Among them, TLR2 and TLR4 have been thought to be crucial for the recognition of common pathogenic fungi, including Candida and Aspergillus species. In particular, macrophages lacking a functional TLR4 or both TLR2 and TLR4 result in decreased nuclear translocation of NF-κB and release of various pro-inflammatory cytokines in response to viable conidia of Aspergillus fumigatus.37 In addition, the lack of TLR2 and/or TLR4 significantly impairs recruitment and effector function of neutrophils in response to Aspergillus fumigatus. Furthermore decreased recruitment of neutrophils and macrophages to the site of Candida albicans infection was observed in TLR2−/− mice,38 and mice expressing nonfunctional TLR4 showed decreased phagocytosis and production of pro-inflammatory mediators in macrophages against Candida albicans infection.39
NLRs
NLRs, a cytoplasmic PRR family, are also involved in the recognition of fungi and induction of anti-fungal immune response.4041 Two NLRs, namely, NLRP3 and NLRC4, are reported to sense fungi to form the proteolytic multiprotein complex termed inflammasomes, processing and activating IL-1β and IL-18 which are essential cytokines for protection against fungi.2941 On sensing certain fungi, these NLRs oligomerize to form caspase-1-activating scaffold through recruiting and nucleating numerous apoptosis-associated speck-like proteins containing a CARD (ASCs) and pro-caspase-1. Then, active caspase-1 cleaves pro-IL-1β and pro-IL-18 into their biologically active forms IL-1β and IL-18, respectively. Interestingly, activation of NLRP3 inflammasome against Aspergillus fumigatus and Candida albicans may require Syk tyrosine kinase, which is located in the downstream of CLR signaling.40 Furthermore, the CLRs-Syk pathway is also known to be related to non-NLR-associated inflammasome to produce IL-1β against fungi.29 Therefore, the CLRs-Syk pathway may be a potent upstream modulator in the activation of various inflammasomes against fungi.
Action of fungal proteases
Many of the fungal allergens are known as proteases (www.allergen.org. Accessed July, 2017). Similarly, increased expression of protease-activated receptor-2 (PAR-2) is observed in asthmatic airway epithelium and diverse proteases from many aeroallergens, including fungi, activate PAR-2.42 Therefore, elucidating the action mechanism of various fungal proteases in allergic lung disease is an important issue to unveil the fundamental molecular basis of the disease. In particular, Homma et al.43 recently demonstrated that PAR-2 activation by Aspergillus fumigatus may weaken TH1 cells-mediated antiviral/interferon response without affecting Th2 cells-mediated response in airway epithelial cells, and thus this may impact on airway cell populations toward TH2 cells. Additionally, highly purified protease derived from Aspergillus fumigatus was shown to be sufficient to convert type I allergen ovalbumin (which typically require aluminum-based adjuvants before respiratory challenge to induce pulmonary allergic inflammation in mice) to type II allergen (which can readily induce allergic lung inflammation via airway challenge without additional extra-pulmonary adjuvants) when primary respiratory exposure to ovalbumin occurs in the presence of fungal protease, which implies that active proteases are the essential adjuvant factor responsible for overcoming tolerogenic responses and inducing allergic reaction.44 Furthermore, fibrinogen cleavage product by fungal protease in airway epithelium has been reported to activate PRRs, such as TLR4, also known as one of the important upstream regulators of T helper responses to allergens,4546 thereby associating antifungal innate immunity with adaptive allergic inflammation.13 Importantly, fungal proteases directly influence the integrity of asthmatic bronchial epithelium and induce morphologic changes and pro-inflammatory cytokine production, thereby possibly enhancing the allergenic potential of the other fungal allergens at the same time.7 Given that epithelial exposure to various fungal allergens occurs simultaneously, these aspects of action may highlight the vital role of fungal proteases in initiating allergic airway disorders.
Interpretation of fungal exposure and induction of host immune responses
Initial fungal recognition leads to release of endogenous danger molecules, including uric acid, ATP, and various epithelium-derived cytokines, such as IL-33, IL-25, and thymic stromal lymphopoietin (TSLP) from airway epithelium.45 These molecules are critically implicated in the pathogenesis of fungal allergy. For example, IL-33 is a member of the IL-1 family of cytokines and normally localized in the nucleus of airway epithelial cells. However, IL-33 can be released and processed to a potent mature form in response to epithelial stimulation from protease action and PRR activation. Then, through engagement of IL-33 with IL-1 receptor family member suppressor of tumorigenicity 2 (ST2), group 2 innate lymphoid cells (ILCs) can be activated and rapidly induce type 2 inflammation in the airways. In this context, IL-33/ST2 signaling has reported to be prerequisite for airway eosinophilia following exposure to Alternaria alternata, which is mainly orchestrated by IL-5/IL-13-producing group 2 ILCs in the lung.47 Moreover, type 2 airway inflammation induced by chitin (a polysaccharide constituent of fungal cell wall) and protease allergen papain also involves epithelial production of IL-33, IL-25, and TSLP, and subsequent activation of group 2 ILCs, DCs, and other innate immune cells.4849 Importantly, epithelial release of IL-33 leads to exacerbation and airway remodeling of allergic airway disease in murine models,5051 which highlights the possible implication of IL-33 in fungi-induced severe allergic inflammation. Furthermore, a recent study has shown that children with SAFS is associated with higher airway IL-33 levels in bronchoalveolar lavage fluid and endobronchial biopsy specimens compared to children having severe asthma without fungal sensitization. Similarly, pulmonary IL-33 levels and IL-13+ group 2 ILCs numbers are increased in the Alternaria alternata-induced SAFS animal model. Notably, Alternaria alternata-induced elevations in lung IL-33 levels and IL-13+ group 2 ILCs numbers remain increased with CS treatment, demonstrating that anti-fungal host immune response involving epithelial IL-33/group 2 ILCs may contribute to CS resistance of fungi-induced allergic lung inflammation.52
Genetic background of the host
Several genetic problems affecting the phagocytic activity of cell-mediated immunity have been well known to be associated with increased risk of fungal infection.53 For instance, genetic defects in several proteins comprising the NADPH oxidase complex, which is essential for the generation of reactive oxygen species (ROS) in phagocytes, are related to increased incidence of invasive fungal infection. However, scarce information is available on genetic associations concerning SAFS and other fungal allergic diseases. Most knowledge on these subjects came from small studies on ABPA. Several implicated genes have been identified, which includes IL-4Rα, cystic fibrosis transmembrane conductance regulator, and HLA-DR.26 In regards to SAFS, HLA class II restriction involving HLA-DR and HLA-DQ was reported to be implicated in Alternaria-sensitive moderate-to-severe asthma.54 In particular, there is possibility that genetic loci encoding IL-33 and its receptor could be linked to SAFS development, since these have consistently been reported to be a critical susceptibility factor for human asthma.55 Further research is warranted regarding this issue.
Implication of ER Stress in Fungal Allergy and SAFS
ER stress and unfolded-protein response (UPR)
ER is located adjacent to nuclear envelope and structurally consists of continuous membrane network that expands throughout the cytoplasm. ER participates in a variety of cellular functions. In addition to its well-known role as a major cellular storage for calcium, ER plays an essential role in the synthesis, correct folding, assembly, modification, and transport of soluble and membrane proteins. To efficiently accomplish its roles as a ‘protein folding factory,’ ER is abundantly equipped with chaperones, oxidases, and isomerases that facilitate proper folding of proteins and thus manipulates nearly one-third of the cellular proteome.18
Meanwhile, optimal milieus within ER, such as adequate concentrations of ATP and calcium or oxidizing environments, are essential to retain the maximal activities of many enzymes for protein folding.56 Therefore, ER is highly sensitive to various stresses that perturb cellular energy levels, calcium concentration, and redox state, all of which can lead to the accumulation of misfolded and/or unfolded proteins in the ER lumen. Accumulation of misfolded/unfolded proteins are toxic to cells and can cause an imbalance between the ER folding capacity and the folding load of nascent protein entering ER, which is referred to as ER stress. To maintain cellular homeostasis against this crisis, eukaryotic cells have evolved adaptive response, UPR. UPR signaling is mediated by 3 ER-localized sensors in mammalian cells. These consist of inositol-requiring 1α (IRE1α), double-stranded RNA-dependent protein kinase (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6); each of these ER transmembrane proteins senses unfolded proteins in ER through ER-luminal domain. According to the proposed model of UPR activation, all these sensors remain in the inactive state through association with abundant ER chaperone glucose-regulated protein 78 (GRP78) in resting cells. In a condition of ER stress, however, GRP78 preferentially associates with accumulated unfolded proteins in ER and dissociates from the 3 sensor proteins. Then, ER stress sensor proteins become activated (as for IRE1α, direct activation through engagement with misfolded proteins has been also demonstrated)57 and transmit signals about the folding status of ER to the cytosol and nucleus, inducing UPR-target genes to restore the ER folding capacity.58 Although the 3 UPR signaling pathways are simultaneously activated in response to the accumulation of unfolded/misfolded proteins, contribution of each pathway substantially varies depending on the severity and chronicity of ER stress.
Activation of the PERK pathway is known to be a relatively immediate response to ER stress. PERK is a serine/threonine protein kinase and, on its activation, PERK becomes dimerized and autophosphorylated. This process allows PERK to phosphorylate the α-subunit of eukaryotic translation-initiation factor 2α (eIF2α), which inhibits the assembly of the 80S ribosome in eukaryotic cells, therefore the synthesis of proteins. Considering that blockade of PERK-induced eIF2α phosphorylation leads to abnormally elevated protein synthesis and higher levels of ER stress, this pathway may reduce ER protein folding load in ER stressed-cells. Furthermore, eIF2α phosphorylation stimulates translation of selective mRNAs, including ATF4, that contain regulatory sequences, such as an inhibitory upstream open reading frame in 5′-untranslated regions. ATF4 is a transcription factor that has been known to control pro-survival genes in relation to protein folding, anti-oxidant response, and ER-stress-induced apoptosis (apoptosis occurs later during ER stress when the other arms of UPR fail to restore equilibrium).59
Cleavage of ATF6 follows relatively quickly in response to ER stress. When ATF6 is released from GRP78, it moves to the Golgi apparatus where it is cleaved. This process causes the release of a functional cytosolic fragment of ATF6 (ATF6f). ATF6f translocates to the nucleus and induces gene transcriptions of enzymes that help folding, maturation, secretion, and ER-associated degradation (ERAD) of protein. ATF6 seems to modulate a relatively narrow range of genes participating in the ER protein quality control system, many of which are also influenced by the IRE1α/X-box-binding protein 1 (XBP1) pathway. This may be why mice lacking ATF6 are overtly normal with no apparent phenotype. Thus, the ATF6 pathway seems to fine-tune and enhance the functional capacity of UPR along with PERK and IRE1α, especially in chronic ER stress.6061
The IRE1α pathway is the most evolutionarily conserved one among 3 major branches of UPR. IRE1α has both serine/threonine protein kinase activity and site-specific endoribonuclease (RNase) activity. GRP78 dissociation from IRE1α or direct engagement of IRE1α with misfolded proteins promotes IRE1α dimerization and autophosphorylation, activating its RNase activity. IRE1α catalyzes the splicing of mRNA encoding XBP1 and generates a spliced variant (sXBP1) that functions as a transcription factor for genes associated with the structural and functional expansion of ER. Alone or with ATF6, sXBP1 induces chaperones, such as GRP78 and proteins, participating in ER biogenesis, lipid synthesis, ERAD, and protein secretion.58 Because the synthesis of XBP1 mRNA is up-regulated by ATF6 following ER stress, full activation of the IRE1α-XBP1 pathway may be delayed compared to the other pathways of UPR.56 However, transcriptional targets of XBP1 are not just limited to the restoration of the ER protein quality control system. The IRE1α/XBP1 pathway also participates in the induction of molecules associated with lipid metabolism,62 immune and inflammatory responses,63 and cellular differentiation,64 so that it plays previously unrecognized roles in many critical cellular events. Consistent with these findings, mice lacking XBP1 display hypoplastic fetal livers leading to the death from anemia65 and XBP1 is also known to be important in the terminal differentiation of B cells into highly secretory plasma cells.66 Moreover, homozygous ire1α−/− embryos show lethal defects in the differentiation of liver and B lymphocytes.67 Furthermore, in contrast to the specific endoribonuclease activity against XBP1, IRE1α also has non-specific RNase activity and may degrade ER-membrane-associated mRNAs to reduce the production of proteins, namely, regulated IRE1α-dependent decay (RIDD).68 This cellular mechanism is well suited to complement other UPR pathways. Recently, increasing evidence has indicated that expression of a wide range of proteins, involved in diverse cellular processes beyond ER homeostasis, can be regulated through RIDD, explaining the critical involvements of UPR pathways in broad cellular events other than cellular protein homeostasis.69
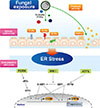
Cumulatively, these canonical aspects of UPR generally attempt to reduce ER stress by reducing the demand of protein folding, facilitating the protein degradation pathway, and increasing the expression of enzymes and ER chaperones that help in protein folding. If causative stress is very severe and the cell fails to resolve the protein folding defect, then these adaptive responses will initiate apoptosis. CCAAT/enhancer-binding protein homologous protein (CHOP), as a downstream effector of UPR, is known to mediate ER-stress-induced apoptosis. Thus, CHOP, along with GRP78, is widely used as a marker which indicates the presence of ER stress2125 and up-regulation of GRP78 is typically detectable earlier than CHOP (Fig. 1).
Non-canonical aspects of UPR
More recently, there is accumulating evidence suggesting that ER stress and UPR pathways are closely cross-linked to diverse signaling outcomes having seemingly little to do with ER function as a protein folding factory. Indeed, our knowledge has expanded that there are miscellaneous non-canonical aspects of UPR providing mechanistic insight into the pathogenesis of various human inflammatory diseases. As comprehensively reviewed by Arensdorf and colleagues,18 non-canonical UPR can arise from multiple points of canonical UPR through various mechanisms.
First of all, canonical UPR can result in translational regulation of specific proteins particularly through the PERK-eIF2α pathway. Since phosphorylation of eIF2α transiently suppresses almost 90% of cellular mRNA,70 the PERK pathway can efficiently converge on a wide range of cellular processes, including inflammatory process. Specifically, NF-κB signaling, which is a well-known master transcription factor involved in pro-inflammatory cytokine production, leukocyte recruitment, or cell survival,71 seems to be partly controlled by phosphorylation of eIF2α through suppressing translation of inhibitors of NF-κB (i.e. IκB).72 Moreover, this pathway also enables the translation of a number of transcription factors beyond ATF4.707374
Secondly, the sensor proteins of canonical UPR, including IRE1α and PERK, can create stress-specific scaffolds on ER membrane. IRE1α and PERK have been demonstrated to become self-associated to form high-order oligomerization in cytosolic domains, followed by autophosphorylation through action of the kinase domain.75 Furthermore, high-order assembly of UPR sensor proteins has been proposed to provide a specialized molecular microenvironment, which can be associated with low-affinity binding molecules with relatively high avidity.76 Therefore, this allows an additional control principle in regulating UPR and other signaling pathways. The best example for protein interactions with phosphorylated and oligomerized IRE1α is tumor necrosis factor receptor-associated factor 2 (TRAF2), which is essential for the activation of several protein kinases. The IRE1α-TRAF2 complex recruits and activates c-JUN N terminal kinase (JNK), which induces the expression of genes involved in inflammation partly through phosphorylating the transcription factor activator protein 1.77 In addition, the IRE1α-TRAF2 complex is implicated in the recruitment of IκB kinase, which phosphorylates IκB, resulting in the degradation of IκB and the activation of NF-κB.78 Consistent with these data, in the IRE1α−/− mouse embryonic fibroblasts, decreased activity of JNK against ER stress79 and impairment of ER stress-induced NF-κB activation and subsequent reduced expression of TNF-α78 were observed. Regarding the PERK pathway, there are fewer known molecular interactions arising from stress-specific scaffolds than the IRE1α pathway. It has been demonstrated that PERK is required for the phosphorylation of nuclear factor-erythroid 2-related factor-2 (Nrf-2), a key transcription factor implicated in cellular defense against oxidative insults, and cells harboring a targeted deletion of PERK exhibit failure to upregulate Nrf-2-target genes.80
Thirdly, many of the transcription factors in canonical UPR (e.g. ATF6, ATF4, XBP1) belong to the transcription factor family having basic leucine zipper (bZIP) domain and numerous molecular interactions between members of this family protein have been reported.81 Thus, these interactions among bZIP family members can significantly impact on gene expressions under ER stress. For instance, ATF6 is known to interact with CCAAT/enhancer-binding protein β, a member of the bZIP transcription factor family that participates in diverse cellular physiologies, including proliferation, differentiation, metabolism, and inflammation.82 Furthermore, transcription factors in canonical UPR can affect global gene expression beyond their target-genes via sharing coregulatory molecules with constitutive transcription factors.18 Through this mechanism, ATF6 disrupts the costimulatory interaction between cAMP-responsive element-binding protein (CREB), a member of the bZIP family, and CREB-regulated transcription coactivator 2 during ER stress associated with hepatic gluconeogenesis, inhibiting gluconeogenic program.83
Additionally, the expansion of UPR outcome can be achieved by regulating gene expression through RIDD of several other ER-localized mRNAs by the RNase activity of IRE1α,68 by influencing the transcriptome and accompanying diverse physiologic processes through modulating the expression of multifunctional secondary UPR-regulated transcription factors, such as CHOP,18 and by UPR-mediated regulation of mRNA through short single-stranded microRNAs, which promote the degradation of complementary mRNA.84 Defining the non-canonical involvement of UPR pathways in diverse cellular events is an emerging area of investigation. All of these mechanisms may also explain biological consequences of ER stress and highlight the therapeutic potential of ER stress in a wide array of human pathologic conditions.
ER stress and UPR in the lung
Given that huge amounts of foreign materials are inhaled on a daily basis, recognition of these environmental cues should be accompanied by appropriate cellular response in the lung. Accordingly, close interactions between various cell types, including frontline immune cells and adaptive T and B cells, are essential. Importantly, these processes largely rely on cellular secretory function. Therefore, the functional integrity of ER is vital to maintain lung homeostasis against increasing demands of protein folding and secretion.22 For examples, XBP-1 has been reported to be vital for ER/calcium store expansion and subsequent secretion of inflammatory cytokines in airway epithelium.85 Similarly, TLR signaling-induced activation of the IRE1α/XBP1 pathway in macrophages is required for the production of certain pro-inflammatory cytokines, such as IL-6, IL-8, and TNF.63 Furthermore, the optimal functional competence of ER and UPR pathways are known to be essential in the development and differentiation of eosinophils, DCs, plasma cells, and subsets of T cells, thereby linking basic cell biology to broad immunological outcomes in the lung as reviewed elsewhere.86
Previous research has also highlighted the involvement of ER stress and UPR in numerous pulmonary conditions associated with common environmental insults. For example, inhaled fine particulate matter (aerodynamic diameter <2.5 µm, PM2.5) activates ER stress, (especially the PERK/CHOP pathway) and causes ER stress-associated cellular apoptosis through a ROS-dependent mechanism.87 Additionally, the experimental exposure of cigarette smoke and aqueous extracts of cigarette smoke induces ER stress and PERK-mediated survival UPR activation in addition to direct oxidative effects in human lung epithelial cells88 and mouse fibroblast,89 respectively, which emphasizes the role of UPR in maintaining cellular redox homeostasis against oxidative stress. Moreover, XBP-1 mediates ER/Ca2+ store expansion and potentiates IL-8 secretion during airway inflammation associated with Pseudomonas aeruginosa infection85 and IRE1α and PERK pathways are known to suppress viral protein synthesis under viral infection.90
Role of ER stress in allergic inflammation of SAFS
For years, there has been much progress in our knowledge on the implication of ER stress in inflammatory processes, which has been vastly reviewed elsewhere.1820 As for the respiratory system, ER stress and UPR are closely interconnected with various cellular signaling networks, being involved in numerous inflammatory lung disorders including bronchial asthma.2122232425
Disturbances in ER homeostasis can influence diverse aspects of allergic inflammatory process in the lung. First, many triggers of asthma have also been demonstrated as potent inducers of ER stress and UPR in the lung.22 For instance, TLR4 activation by house dust mites (HDMs) in airway epithelium is known to be essential in allergic lung inflammation,46 and TLR4 signaling and the IRE1α/XBP1 arm of UPR coordinate immune response in the production of certain cytokines, such as IL-6 and TNF, in macrophages.63 Additionally, several proteins related to the structural and functional integrity of ER have been known to be implicated in the development of allergic lung inflammation. Reticulon-4 (RTN-4, also known as the Nogo family) is a member of the RTN family of proteins, which is largely restricted to ER and participates in shaping and structuring ER membranes.91 Nogo-B, an isoform of Nogo, is predominantly expressed in the lung and expression of Nogo-B is markedly reduced in a murine model of allergic lung inflammation and human fatal asthmatic lungs. Interestingly, Nogo-knockout mice display exaggerated allergic lung inflammation and epithelial reconstitution of Nogo-B attenuates asthma-like phenotypes in these mice.92 Allergen-induced mucin overproduction is one of the hallmarks of bronchial asthma. Given that airway mucins are large (~5,000 amino acid residues) glycoproteins and post-translational modification of immature mucin is accomplished in ER, ER resident protein disulfide isomerase (PDI) anterior gradient homolog 2 (AGR2), which interacts with immature mucin, is likely to be involved in allergic lung inflammation. Consistent with this assumption, Agr−/− mice display ER stress in airway epithelium and impaired mucin production during allergic lung inflammation.93 IRE1β, an isoform of IRE1, mainly expressed in epithelial cells of the gut and the lung, has been reported to be involved in allergen-induced goblet cell differentiation and airway epithelial mucin production partly through XBP-1-dependent transcription of AGR2.94 Notably, a well-known asthma-associated gene, orosomucoid-like 3 (ORMDL3), belongs to ER membrane protein that is involved in membrane biogenesis.95 Moreover, orm1Δ orm2Δ yeast that lacks the yeast members of the ORMDL family displays constitutive UPR and susceptibility to ER stress inducer.96 A previous study also demonstrated that ORMDL3 is an allergen and a TH2 cytokine-inducible gene predominantly expressed in airway epithelium, and it induces expression of various mediators of allergic inflammation and UPR specifically the ATF6 pathway, highlighting a possible mechanistic link between the ER protein quality control system and bronchial asthma.97
Notably, increasing evidence has demonstrated that ER stress may be one of the key players in the development of severe allergic lung inflammation commonly refractory to conventional treatment, such as inhaled/systemic CS.98 One important study demonstrated the critical involvement of ER stress in bronchial asthma, especially neutrophilic asthma which commonly manifests the severe disease phenotype having CS resistance.21 In that study, representative ER stress markers, including GRP78 and CHOP, in the lung were increased in a murine model of neutrophilic asthma and asthmatic patients. Interestingly, inhibition of ER stress using a chemical chaperone, 4-phenylbutyric acid (4-PBA), resulted in the marked improvement of neutrophilic allergic lung inflammation and the reduction in protein levels of ER stress and UPR markers, including ATF6α, XBP-1, and p-eIF2α. However, dexamethasone treatment failed to decrease neither neutrophilic allergic lung inflammation nor elevation of ER stress or UPR proteins. In another study, allergeninduced airway fibrosis, a critical manifestation of severe asthma, was closely associated with ER resident protein 57 (ERp57), an ER-localized chaperone involved in glycoprotein folding and secretion. In that study, levels of ERp57 were predominantly increased in the epithelium of asthmatic patients and murine models of asthma, indicating the increased protein folding load. Notably, allergen-induced increases in collagen and smooth muscle actin, well-known fibrotic markers, were significantly decreased by specific ablation of epithelial ERp57.99
Meanwhile, considering the pivotal role of ER stress in severe asthma, ER stress and UPR pathways may contribute to the pathogenesis of fungi-associated severe allergic lung inflammation, including SAFS (Fig. 1). Although scarce information exists, interesting data from our group has shown the crucial involvement of these pathways in Aspergillus fumigatus-induced allergic inflammation.100 In this study, Aspergillus fumigatus exposure in mice resulted in characteristic features of fungi-induced allergic lung inflammation—including elevated pulmonary TH2-associated cytokines, such as IL-4, IL-5, and IL-13, total and Aspergillus fumigatus-specific IgE in serum, eosinophil-dominant allergic lung inflammation, and bronchial hyper-responsiveness. Interestingly, treatment with dexamethasone failed to improve Aspergillus fumigatus-induced allergic lung inflammation, suggesting that the murine model displays characteristic features of severe fungal allergic inflammation, such as SAFS. Protein levels of ER stress (GRP78 and CHOP) and UPR-related markers (p-IRE1α, p-eIF2α, XBP-1, and ATF-4) were also remarkably increased in the lung of Aspergillus fumigatus-exposed mice, and increases in GRP78 were observed in the lung tissues samples from patients with ABPA. Importantly, administration of 4-PBA markedly attenuated Aspergillus fumigatus-induced ER stress and improved characteristic features of Aspergillus fumigatus-induced allergic lung inflammation, while dexamethasone treatment did not. These findings suggest that ER stress may play a key role in the pathogenesis of fungal allergic lung inflammation, especially in the manifestation of severe phenotypes of the disease.100
Currently, it is not known exactly on the mechanism through which airway fungal exposure specifically induces eosinophilic allergic inflammation. However, one possible explanation would be related to NF-κB activation, a well-known master regulator of allergic inflammation. It has been demonstrated that close associations of ER stress and UPR with NF-κB signaling play a key role in ER stress-related inflammatory processes, specifically in lung inflammation.2125 Similarly, nuclear translocation of NF-κB p65 was remarkably increased in the lung tissues from the murine model of Aspergillus fumigatus-induced fungal allergic lung inflammation.100 In addition, respiratory exposure to Aspergillus fumigatus resulted in increases in pulmonary levels of type 2 cytokines, such as IL-4, IL-5, and IL-13, leading to eosinophilic lung inflammation. Importantly, treatment with an inhibitor of NF-κB into mice significantly reduced Aspergillus fumigatus-induced increases in the levels of type 2 cytokines and eosinophilic allergic inflammation.100 These findings suggest the crucial implication of NF-κB signaling in fungi-induced eosinophilic allergic lung inflammation.
In addition, it seems that several cross-talks between ER stress and other cellular inflammatory signaling platforms exist, all of which can further explain the molecular mechanism of severe allergic lung inflammation associated with fungi. These include phosphoinositide 3-kinase-δ (PI3K-δ), mitochondrial ROS (mtROS), and NLRP3 inflammasome.
Cross-talks between ER stress and cellular inflammatory signaling platforms leading to severe allergic inflammation in SAFS
PI3K-δ
PI3Ks are lipid signaling kinases that control a variety of crucial cellular events. Class I PI3Ks are generally associated with cell membrane receptors, such as growth factor and cytokine receptors, and phosphorylate the 3′-position of inositol lipids to generate second messenger phosphatidylinositol-3, 4, 5-trisphosphate at the cell membrane, which serves as a cellular docking molecular platform for proteins possessing pleckstrin-homology-domain-containing proteins, such as AKT.101 This process leads to downstream cascades of protein-protein interactions and phosphorylation, culminating in multiple biological consequences. Class I PI3Ks consist of four members (namely, PI3K-α, PI3K-β, PI3K-γ, and PI3K-δ), all of which are heterodimeric complexes having a catalytic p110 subunit (α, β, γ, or δ) in association with a regulatory subunit. While expressions of p110-α and p110-β isoforms are ubiquitous in most cell types, expressions of p110-γ and p110-δ isoforms are restricted to circulating leukocytes and both isoforms have been reported to play key roles in leukocyte signaling.102 Particularly, PI3K-δ plays specific roles in a variety of immunologic processes involving antigen receptor signaling in T cells and B cells, mast cell degranulation, and the migration and activation of neutrophils and eosinophils. The crucial role of PI3Ks, especially PI3K-δ isoform in allergic lung inflammation has also been uncovered.103104 Blockade of PI3K-δ has also shown to attenuate CS-resistant severe inflammatory processes, including severe asthma and COPD.100105 Therefore, inhibition of PI3K-δ is regarded as a promising target for developing a novel drug for chronic inflammatory airway disorders, especially for CS-resistant inflammation.
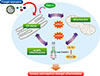
PI3K pathways seem to be closely associated with ER stress and UPR. One report demonstrated that regulatory subunits of PI3Ks (p85) participate in the translocation and possibly stabilization of XBP-1, and that disruption of this interaction results in a severe defect in lowering ER stress.106 In addition, inhibition of PERK leads to decreased activation of PI3K/AKT signaling through regulating cellular localization of PTEN, a negative regulator of the PI3K pathway.107 Furthermore, ER stress-induced Sirtuin 1 (SIRT1) expression, which is involved in ER stress-induced damage responses, has been shown to be regulated by the PI3K-AKT signaling pathway.108 Remarkably, we recently demonstrated the critical involvement of PI3K-δ in the regulation of ER stress in a murine model of SAFS (Fig. 2).100 In this study, the blockade of PI3K-δ ameliorated Aspergillus fumigatus-induced severe eosinophilic allergic lung inflammation that was refractory to dexamethasone. Furthermore, inhibition of PI3K-δ significantly reduced Aspergillus fumigatus-induced increases in ER stress and UPR markers, particularly in airway epithelial cells. Eventually, therapeutic effects of PI3K-δ blockade were closely linked to the attenuation of ER stress-associated NF-κB activation. Taken together, an important cross-talk between ER stress and PI3K-δ is likely to be implicated in the pathogenesis of SAFS.
mtROS
Oxidative stress has long been proposed as one of the essential features in chronic airway disorders.109 Exposure to various oxidants has been reported to cause several cardinal features of asthma, including allergic airway inflammation and airway hyper-responsiveness (AHR).110111 In the same manner, the functional incompetence of cellular anti-oxidant systems is also known to be closely implicated in the pathogenesis of bronchial asthma through inducing oxidative stress. ROS can impact on numerous aspects of the inflammatory process in the lung. First, ROS can activate a wide range of cellular signaling by themselves. ROS also interact with diverse biomolecules, such as lipids and proteins, thereby producing secondary mediators which possess a wide range of effects in the body. Moreover, ROS can directly cause protein modification and DNA damage. Furthermore, oxidative stress can be even more intensified through the recruitment of diverse inflammatory cells, another important endogenous source of ROS, to the lung.109
Given that quantities of ROS are vital for determining its specificity and function and that mitochondria are regarded as the most powerful source of intracellular ROS, mtROS should be tightly regulated in cells. Numerous factors can regulate mtROS in the processes of generation or elimination.112 Generation of mtROS can be controlled by various cellular stimuli, including increased cytosolic Ca2+ concentration, activation of cellular signaling pathways involving immunoreceptors and cytokines, redox status of electron transport chain (ETC), and electrical gradient in inner mitochondrial membrane.112113 As for the removal of mtROS, several essential antioxidant systems, including glutathione peroxidases, peroxiredoxins, and catalase, can eliminate overproduced mtROS.
Furthermore, production of ROS and subsequent development of oxidative stress are known to be important in modulating the protein-folding capacity of ER.114115 In particular, ER stress and mtROS can be interconnected, affecting both aspects of mtROS regulation in the cell. Previous reports have shown that increased leak of Ca2+ from the ER lumen in response to ER stress or cellular oxidative stress can influence the generation of mtROS from ETC as a consequence of Ca2+ accumulation in the mitochondria.112116 Then, increased generation of mtROS further impact on the Ca2+ release channel in the ER membrane,117 thereby increasing Ca2+-release from ER. Furthermore, decreased mitochondrial functional integrities result in release of more antioxidant enzyme, such as glutathione in the mitochondrial matrix as well, perpetuating oxidative stress as a vicious cycle.112114
Although the precise role of mtROS is not fully understood in allergic lung inflammation, a recent study showed that mtROS generation against common allergens is important in allergic inflammation, especially in airway structural cells.118 In that study, increased mtROS generation was observed in airway inflammatory cells and tracheal epithelial cells from ovalbumin (OVA)/lipopolysaccharide (LPS)-sensitized and OVA-challenged mice which showed neutrophil-dominant allergic inflammation. Interestingly, treatment with a potent mtROS scavenger, NecroX compound,119 reduced allergen-induced mtROS generation in these cells and ameliorated various features of allergic lung inflammation through modulating NLRP3 inflammasome activation in airway epithelial cells. Furthermore, the therapeutic effect of mtROS scavenger was also demonstrated in another murine model of allergic lung inflammation induced by HDMs.
Importantly, mtROS may also be closely interconnected with ER stress in the pathogenesis of fungi-induced severe allergic lung inflammation.100 In that study, increased production of mtROS was observed in airway inflammatory cells and tracheal epithelial cells from Aspergillus fumigatus-sensitized/challenged mice. Moreover, Aspergillus fumigatus-exposed mice showed decreased glutathione and increased glutathione disulfide levels in the lung compared to the control mice, which indicates the presence of oxidative stress. Treatment with a mtROS scavenger improved various features of Aspergillus fumigatus-induced severe eosinophilic allergic lung inflammation, and a mtROS scavenger also lowered Aspergillus fumigatus-induced elevations of ER stress markers in the lung. These results suggest that mtROS contribute to the modulation of ER stress in fungal allergic inflammation, and that this crosstalk may be involved in the development of SAFS (Fig. 2).
NLRP3 inflammasome
NLRP3 inflammasome has been reported to be essential in anti-fungal immune response.40 However, the role of NLRP3 inflammasome in the pathogenesis of fungus-induced allergic lung inflammation remains poorly understood. Considering that ER stress can induce the release of diverse damage-associated molecular patterns (DAMPs) from mitochondria (e.g. mtROS, mitochondrial DNA [mtDNA], ATP, calcium), which are potent activators of NLRP3 inflammasome in the cytosol,120 interconnections between NLRP3 inflammasome and ER stress may play a role in the pathogenesis of fungi-induced allergic lung inflammation. Furthermore, because NLRP3 inflammasome has been reported to be associated with CS-resistant inflammation in the lung,118121 ER-NLRP3 inflammasome interactions are likely to be involved in the development of SAFS.
Interestingly, in our unpublished data, exposure to Aspergillus fumigatus resulted in NLRP3 inflammasome activation in the lung of mice, especially in bronchial epithelial cells. Expression of NLRP3 in the lung tissues from patients with ABPA was also increased. Furthermore, a selective inhibitor of NLRP3 inflammasome122 reversed Aspergillus fumigatus-induced increases in IL-1β protein in the lung of mice, and treatment with IL-1β-neutralizing antibody dramatically attenuated Aspergillus fumigatus-induced severe eosinophilic allergic lung inflammation. Notably, a mtROS scavenger also reversed the Aspergillus fumigatus-induced increases in IL-1β and attenuated characteristic features of Aspergillus fumigatus-induced severe inflammation. These results suggest that NLRP3 inflammasome in association with mtROS/ER stress may be one of the pivotal players in the development of SAFS (Fig. 2).
CONCLUSIONS
Previously unappreciated roles of fungi in bronchial asthma, especially in a more severe phenotype of the disease, have been increasingly demonstrated through numerous epidemiologic and translational studies. However, much knowledge on this field is unanswered so far. Recently, SAFS has been proposed as one of the fungal sensitization/allergy development-associated clinical disease entities to further investigate fungi-associated severe asthma phenotypes. Importantly, recent advances in our knowledge that intracellular organellar stress responses are closely implicated in the development of SAFS have broadened our understandings on this issue. ER stress and UPR are closely implicated in fungi-induced severe allergic lung inflammation, and amelioration of ER stress, especially in epithelial cell layer, may have the potential for treating the disease. More importantly, intracellular cross-talks between ER stress and other inflammatory signaling platforms may provide some clues for the vital role of fungi on severe allergic inflammation in humans. Further investigations on the interconnections between these stress responses and the other potential key mediators of CS resistance will expand our knowledge on SAFS in the future. Finally, efficacies of treatment strategies targeting ER-associated molecular networks in SAFS need to be thoroughly evaluated through well-designed clinical trials.

 XML Download
XML Download