

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Dear Editor:
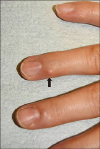
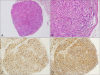
A 45-year-old woman presented with a skin-colored nodule on the right fourth finger that appeared 3 years ago. The nodule had been slowly increasing without any accompanying symptoms. She denied having any past medical history or familial history. Examination of the skin on the right fourth finger revealed a 4×4 mm, firm, nontender, skin-colored, round-shaped nodule (Fig. 1). The lesion was punched out for skin biopsy and pathological analysis demonstrated a relatively well-demarcated dermal nodular mass with plexiform pattern. The mass was mainly composed of histiocytes surrounded by spindle cells, which appeared to be fibroblasts (Fig. 2A, B). The mass had a plexiform and biphasic pattern. Multinucleated, osteoclast-like giant cells were also observed. Immunohistochemistry revealed a strong positivity for CD68 in the histiocytes and vimentin in the fibroblast-like cells, with a faint positivity for smooth muscle actin (SMA) (Fig. 2C, D). Based on this information, the patient was diagnosed with plexiform fibrohistiocytic tumor (PFHT). Following the biopsy, the patient planned to further work-up for rule out the metastases. However, after 6 months, the patient refused to come again due to no further recurrence and no symptom.
PFHT is an intermediate malignant mesenchymal neoplasm, typically presenting in dermal or subcutaneous tissues1. It was named a distinct entity by Enzinger in 198823. He described PFHT as a neoplasm that mainly occurs in children and young adults2. Clinically, it shows female predominance and presents as a solitary, painless, and slow-growing nodule. Most lesions are located on the upper extremities, usually the hand and fingers, but often on the head and neck13. The median age at presentation is 14.5 years (range, 2 months to 71 years)24. Although the pathogenesis of PFHT is not fully understood, dermal infiltration of periostin and CD163+CD206-tumor-associated macrophages are significantly remarkable, as they may be diagnostic markers and may help identify the immunological background of PFHT5. Histopathological features usually include a poorly-demarcated dermal or subcutaneous mass with multinodular plexiform proliferation of fibrohistiocytic cells. Fascicles of fibroblast-like spindle cells surround the nodules of the histiocytes, making a characteristic biphasic pattern. Such neoplastic masses are predominantly located in the subcutis35. Sometimes, osteoclast-like giant cells are also visible. Using these features, PFHT can be histopathologically classified into three types according to the predominant cell type: histiocytic, fibroblastic, and mixed134. The histological appearance of PFHT may be similar to that of plexiform neurofibroma, cellular neurothekeoma, soft tissue giant cell tumor, or dermatofibroma34. The tumor cells are immunohistochemically positive for CD68, vimentin, and SMA. Complete surgical resection is recommended for treatment. As PFHT shows frequent local recurrence (in up to 40% of the cases) and lymph node metastasis (in up to 6% of the cases) after surgical excision145, clinicians should help patients with long-term clinical follow-up3.
To the best of our knowledge, only one case of PFHT in the popliteal fossa in a 4-year-old girl has been reported in Korean dermatologic literature. Our case is specialized, as PFHT presents as a relatively well-demarcated mass in a middle-aged woman. Thus, dermatologists should keep in mind that PFHT could be a possible diagnosis when a single nodule appears on finger, despite having atypical clinicopathological aspects.
 XML Download
XML Download