

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Atopic dermatitis (AD) is a chronic pruritic skin disease characterized by Th2-dominant inflammation including elevated serum immunoglobulin E (IgE) levels and peripheral eosinophilia, as well as abnormal barrier function. AD is one of the most common diseases in childhood and has a complex etiology involving genetic and environmental factors.
Family history has been reported as a risk factor for AD in large population studies1. Many other common and complex disorders have strong genetic heritable components as well2.
AD affects as many as 20% of children in developed countries3, and various mutations of the filaggrin gene have been detected in about 30% of East Asian AD patients4. Filaggrin mutations can predict the severity of AD5. Thus, a broad understanding of the genetic background of this disease is needed for early diagnosis of AD, and for discovering specific therapies that will prevent the development of AD. Identification of genetic variants associated with common diseases and complex traits is required for early detection of AD.
Whole-exome sequencing (WES) has the attractive cost of sequencing better than next-generation sequencing, and can identify functional variants targeted to the exome, which is comprised of about 180,000 exons. Over 85% of pathogenic mutations occur in the protein-coding loci of genes6. WES has already been used to search and annotate variants through multiple analysis of a filtering and prioritization process against reference sequences of all protein-coding regions. Many disease-causing mutations are inherited in a Mendelian, common or complex disorders7.
In an attempt to identify potential causative variants of early-onset familial AD, three pedigrees with AD family history and severe clinical phenotypes were recruited. Functional and large conserved mutations were extracted by WES. A significant number of risk variants were found, and disease-related single-nucleotide polymorphisms (SNPs) were inferred through a literature search.
There is a high probability of variation at evolutionarily conserved amino acid sites having a substantial effect on protein function8. Risk variants extracted via WES were validated in a population-based case-control study using Sanger sequencing, which selectively analyzes sequence regions of interest.
This study suggests a CDKAL1 polymorphism as a novel candidate for the detection of early-onset AD.
MATERIALS AND METHODS
Patients
This study was approved by the Chung-Ang University Hospital Institutional Review Board (IRB no. C2015258 (1716), and involved a family group consisting of two affected and two unaffected individuals respectively (Supplementary Fig. 1). Informed consent was submitted by all subjects when they were enrolled. Each family member developed AD before age two and was selected based on SCOring Atopic Dermatitis (SCORAD) score index (>50) and total serum IgE level (>1,000 kU/L).
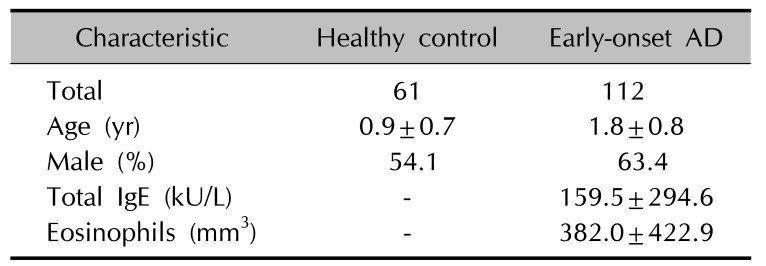
One hundred twelve AD patients and 61 healthy controls under age 2 years 9 months were enrolled, and the characteristics of the study cohort are presented in Table 1. To validate the risk of AD, Sanger sequencing was performed for ERBB2 and CDKAL1 polymorphisms.
Peripheral blood samples were obtained from all subjects. We tried to eliminate environmental factors as much as possible by recruiting early-onset cases and confirming risk variants that might affect heredity. All subjects were diagnosed with AD by a dermatologist.
Whole-exome sequencing
Blood samples were collected in ethylenediaminetetraacetic acid tubes from all subjects, and genomic DNA was isolated using a QIAamp DNA Mini Kit (Qiagen Inc., Valencia, CA, USA). The DNA purity and concentration were measured using a Nanodrop spectrometer (Nanodrop Technologies, Wilmington, DE, USA) and a Qubit fluorometer (Life Technologies, Grand Island, NY, USA). WES was performed using SureSelect Human All Exon V4+UTR 71 Mb (Agilent, Santa Clara, CA, USA), according to the manufacturer's standard protocol. DNA samples were sheared using Covaris (Covaris, Woburn, MA, USA). A paired-end (PE) DNA sequencing library was prepared through shearing, end-repair, A-tailing, peak detection, PE adaptor ligation, and amplification. After the library was hybridized with bait sequences for 24 hours, the captured library was purified and amplified with an index barcode tag. Then, library quality and quantity were measured. Sequencing of the exome library was carried out using the 100-bp PE mode of the HiSeq SBS kit (Illumina, San Diego, CA, USA).
Whole-exome sequencing processing and alignment
Millions of sequence reads in FASTQ format were mapped to UCSC hg19 of the human assembly using the Burrows-Wheeler Aligner (BWA,v0.7.7)9 with “mem” and seed value parameters “−k 45” to create sequence alignment map (SAM) files with correct mate-pair information for the genome. Picard (v1.92) was then used to convert the SAM files to compressed binary alignment map (BAM) files and then to sort the BAM files by chromosome coordinates. The Genome Analysis Toolkit (v2.3.9Lite)10 was used for local realignment process of the BAM files at intervals corresponding to potential insertion and deletion alignment errors. SNPs and indels were annotated using snpEff (v3.6c)11, which classified the variants.
Annotation
The dbNSFP is an integrated database of functional predictions from multiple algorithms (scale invariant feature transform [SIFT], Polyphen2, phylogenetic p-values [phyloP], PhastCons, 1000 Genome).
Filter 1: SnpEff (http://snpeff.sourceforge.net/index.html) annotates type of variants and predicts their effects on genomic regions (e.g., synonymous, nonsynonymous, missense, nonsense, stop-gain, and frameshift point mutations and indels).
Filter 2: SnpEff represents putative variant impact (High:Splice_Site, Start_Lost, Frame_Shift, and Stop_Gained; Moderate: Non_Synonymous_coding, Codon_Insertion and _Deletion; Low: Start_gained, etc.).
Filter 3: SIFT and Polyphen2
SIFT score predicts whether the substitution of amino acids affects protein function.
SIFT prediction is on the basis of the degree of amino acid conservation derived from related sequences through PSI-BLAST. It ranges from 0 to 1; a value lower than 0.05 or not significant is considered detrimental, otherwise it is considered tolerable.
The Polyphen2 HDIV score based on HumDiv, i.e., hdiv_prob ranged from 0 to 1. The scores of prediction were “probably damaging, 0.957~1”; “possibly damaging, 0.453~0.956”; and “benign, 0~0.452.”
Filter 4: The phyloP of dbNSFP reflects the score of evolutionary conservation sites under negative or positive selection over the branches of the phylogenetic tree. A higher score indicates a more conserved site (http://varianttools.sourceforge.net/Annotation/DbNSFP).
Filter 5: PhastCons scores contain multiple alignments of 99 vertebrate genomes to the human genome and predict conserved elements. A high score (>0.2) represents a functionally important genomic region.
Filter 6: The 1000 Genome Project selects variants with minor allele frequencies (MAFs, less than 0.01 or unknown) in the global population.
Filter 7: The in-house Korean variation database at the Theragen Etex Bio Institute selects variants (MAFs, less than 0.02 or unknown).
Sanger sequencing
Polymerase chain reaction (PCR) amplification of two SNPs was performed at 95℃ for 10 minutes, followed by 35 cycles at 95℃ for 30 seconds, 55℃~58℃ for 30 seconds and 72℃ for 40 seconds, with a final extension at 72℃ for 90 seconds. The PCR reaction mixtures (total volume 50 µl) contained 25 µl of 2X EF-Taq premix (SolGent, Seoul, Korea), 18 µl of distilled water, 2.5 µl of oligonucleotide primer (10 pmol/µl), and 2 µl of template containing 20 ng genomic DNA. The PCR products were purified using a PCR purification kit (Favorgen, Pingtung, Taiwan) and were sequenced on an Applied Biosystems 3500 DNA sequencer (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions.
Statistical analysis
Using logistic regression analysis, crude odds ratios (ORs), 95% confidence intervals (CIs), and associations between AD and the SNPs under investigation were calculated. Associations between polymorphisms (rs77152992 and rs1058808) and AD were assessed using Fisher's exact test.
RESULTS
Whole-exome sequencing in three families with atopic dermatitis
We used WES to investigate family-specific candidate variants of three Korean families comprising two AD-affected and two unaffected individuals (Supplementary Fig. 1). To limit our study to genetic influences and minimize environmental factors, we recruited early-onset cases with severe clinical AD phenotypes.
WES was used to sequence the exomes of each individual in three families with a history of AD, and then this variant data was filtered in different steps (Supplementary Fig. 2). To identify susceptible variants for AD, fast and convenient filters for dominant, recessive and heterozygous models were used. A number of variants were excluded in each filtering step, and conserved and functional variants remained in filters 5~7. All variants were counted after each filtering step (Supplementary Table 1,2,3). Considerable numbers of non-synonymous SNPs that affect a codon via amino acid substitution were observed (data not shown). We focused on functional variants which was common variants (MAF greater than 1%) in filter 5, rare variants (MAF lower than 1%) for global population in filter 6 and rare variants (MAF lower than 2%) for Koreans in filter 7.
Papers related to allergic immune diseases were searched using SNPedia (http://www.snpedia.com/index.php/SNPedia) for putative variants of Filter 5~7 (Supplementary Table 1,2,3). We found CDKAL1 (rs77152992) and ERBB2 (rs1058808) non-synonymous SNPs associated with allergic immune diseases like asthma, psoriasis or AD. Although filaggrin SNPs were also detected in WES, they were filtered at a lower step (data not shown).
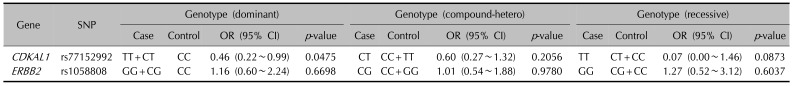
The rs77152992 variant of CDKAL1 was detected in family B through dominant model analysis. All affected individuals were heterozygous for the identified variants, while unaffected individuals were homozygous for the wild-type allele. The rs1058808 variant of ERBB2 was identified in family C through compound-heterozygous model analysis. Both the child and the affected father were heterozygous, while the unaffected mother was homozygous for the alt allele (Supplementary Table 4).
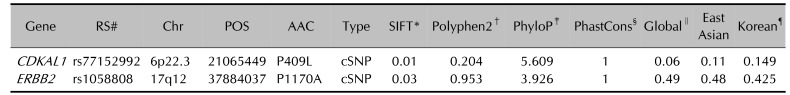
CDKAL1 and ERBB2 SNPs were identified as missense mutations: c.1226C>T, p.Pro409Leu (rs77152992), c.3463 C>G, and p. Pro1170Ala (rs1058808) respectively. rs77152992 was located in chromosome 6p22.3 and rs1058808 was identified in 17q12. The low SIFT scores of the CDKAL1 (0.01) and ERBB2 (0.03) variants predicted high protein damage via amino acid substitution. The high polyphen2 scores of the CDKAL1 (0.204) and ERBB2 (0.953) variants predicted harmful effects on protein structure due to substitution. The phyloP positive scores of the CDKAL1 (5.609) and ERBB2 (3.926) variants indicated that two sites were evolutionarily conserved in multiple alignments of 99 vertebrate genomes to the human genome. The phastcons scores (1) of both sites showed strong conservation in evolutionary events.
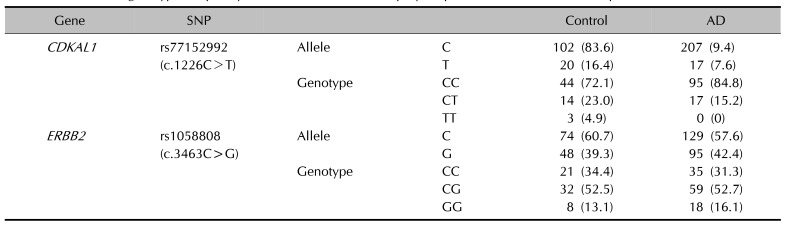
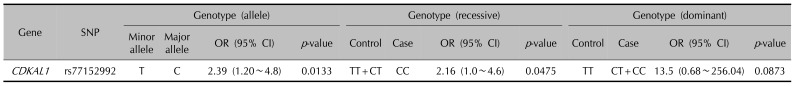
The 6%~14% MAF of the CDKAL1 variant and 43%~49% MAF of the ERBB2 variant were identified in 1,000 global, 286 East Asians, and 800 Koreans, respectively (Table 2). A case-control study was performed to determine whether rs77152992 and rs1058808 are candidate risk factors for early-onset AD. One hundred twelve patients under age 2 years 9 months with early-onset AD and 61 control subjects were enrolled. A total IgE level of 159.5±294.6 KU/L and a total eosinophil level of 382.0±422.9 mm3 for AD patients were used in this study (Table 1). Sanger sequencing was performed, and the allele and genotype frequencies of Koreans were determined by direct gene counting. A distinct reduction in rs77152992 minor alleles (T) was observed in AD patients compared to the control group. The MAF of rs1058808 showed no significant differences between AD and control groups (Table 3).
ORs with 95% CI were estimated for individual risk alleles. rs77152992 of CDKAL1 was significantly associated with early-onset AD (OR, 0.42; 95% CI, 0.21~0.83; p=0.0133) for (T) allele frequency. rs1058808 of ERBB2 had no correlation with AD (Table 4). The TT+CT genotype of CDKAL1 showed a significantly reduced risk of AD development (OR 0.46; 95% CI 0.22~0.99; p=0.0475) under a dominant model (Table 5). However, there was no significant correlation when using a compound-hetero and recessive model. Conversely, the risk of AD was 2.16-fold increase (OR, 2.16; 95% CI, 1.0~4.6; p=0.0475) when major allele CC genotype was set as a recessive deleterious allele (Table 6). ERBB2 showed no association with AD in any of the three models (Table 5).
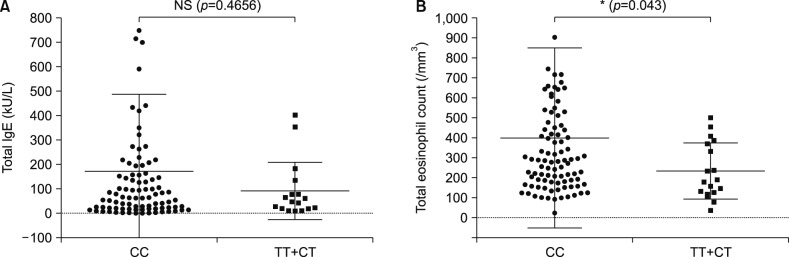
The relationship between the genotype of two SNPs and the clinical characteristics (total IgE level and eosinophil count) of AD patients has been confirmed. Total eosinophil count was significantly increased in AD patients with the CC genotype of CDKAL1 (p=0.043). There was no association between rs77152992 and total IgE levels (Fig. 1). The ERBB2 variant was not related to the clinical characteristics of AD (Supplementary Fig. 3).
DISCUSSION
AD is a common and complex skin disease associated with both genetic and environmental factors. Early development of AD in infancy carries a higher risk for allergic rhinitis and asthma later in life12. In these cases, the detection of risk variants becomes more important for prediction and prevention of AD and allergic march. Loss-of-function mutation in the filaggrin gene, a major structural protein in the epidermis, constitutes a well-recognised susceptibility locus for AD. Further, genome-wide association studies (GWAS), including large meta-analyses, have discovered several additional susceptibility loci with genome-wide significance. However, the reported variations only explain a fraction of the overall heritability of AD13.
To identify candidate variants that may affect disease inheritance, a family-based design was used. We chose a family group consisting of four individuals, where one of the parents and the children developed AD at an early age. The family design was also advantageous due to cost-effectiveness and identification of hidden variants not detectable in a population-based study like GWAS14.
As a method of identifying genetic disease markers, the common disease-common variant (CD-CV) hypothesis was the first dominant theory of common disease in the genetics field. However, the CD-CV hypothesis reached the limit, and the theory that rare variants might have a large effect arose. The issue of which variants have critical effects is still a contentious issue in the field15. In this study, WES found family-specific common and rare variants, yet there were no overlapping mutations that occurred in AD patients compared with healthy controls in three families, despite filtering a massive quantity of variants. We confirmed variants related to allergic disease in each family through a literature search and using three models (dominant, hetero and recessive).
The CDKAL1 gene is a member of the methylthiotransferase family located in 6p22.3. The function of the protein is unknown. The expression of the CDKAL1 gene is absent in primary keratinocytes, whereas abundant expression has been identified in CD4+T cells15. Common SNPs located in the CDKAL1 gene have been linked to increased risk of psoriasis and have been suggested to have an effect on the response to anti-tumor necrosis factor therapies1617. AD also has immune dysregulation mediated by T helper cells, although psoriasis is characterized by a T helper type 1 and/or T helper type 17 immunological response, whereas acute AD lesions exhibit T helper type 2-dominant inflammation18. Functional sequences can be predicted by confirming evolutionary conservation rates across species19. Our data suggest that rs77152992 was a strongly conserved protein coding region among 100 vertebrate species. The similar MAF of rs77152992 between 800 Koreans (14.9%) and 61 healthy controls (16%) demonstrates the credibility of the data despite the small sample size in this case-control study. A previous study reported that a patient with a chromosome 6p22.3 deletion had hypereosinophilic syndrome20. rs77152992 was located in 6p22.3, and a high eosinophil count has been observed in AD patients with major allele (CC) genotype. It was reported that mean level of peripheral blood eosinophil count was 290.0±205.7 total eosinophils/mm3 in AD patients (n=30) and 153.3±113.7 total eosinophils/mm3 in healthy control (n=30)21. Our data showed 400.9±449.6 total eosinophils/mm3 in AD patients with CC genotype and 235.1±140.6 total eosinophils/mm3 in AD patients with CT+TT genotype. The CC genotype of rs77152992 may be associated with increased eosinophil counts. Thus, The CDKAL1 gene and the CC genotype of rs77152992 may be implicated in early-onset AD.
Mean level of total IgE was 224 kU/L (14~12,013 kU/L) in extrinsic AD (n=65) and 25.2 kU/L (0~4,352 kU/L) in intrinsic AD (n=38)22. Our data represented mean level was 172.1 kU/L (2~1,768 kU/L) in AD patients with CC genotype and 91.77 kU/L (10~401 kU/L) in AD patients with CT+TT genotype. There was no correlation between AD patients with CC and CT+TT genotype group.
The ERBB2 gene is a family member of epidermal growth factor receptor. rs1058808 was a candidate causal SNP related to asthma in a GWAS, and transmembrane receptor activity was a candidate casual pathway identified through Identify candidate Causal SNPs and Pathways analysis23. Although rs1058808 is a functional variant with strong evolutionary conservation and can be damaging to proteins, it was not associated with the risk of early-onset AD pathogenesis in a case-control study. Although the rs1058808 variant was not correlated with AD in our data, Sääf et al.24 reported that ERBB2 mRNA was highly expressed during keratinocyte differentiation. This protein level showed lower expression in AD skin compared to healthy controls. Thus, amino acid substitution of rs1058808 may be more likely to impact AD in patients with the mutation.
Taken together, two candidate variants, CDKAL1 (rs77152992) and ERBB2 (rs1058808), were identified in familial genomic data. The high evolutionary conservation of these two SNPs suggested deleterious protein function. Our case-control study suggests that the CC genotype of rs77152992 may be associated with increased eosinophil counts. It may enhance the risk of early-onset AD. Our study strengthens the support for a genetic basis of early-onset Korean AD, and contributes to the development of a diagnostic and predictive tool for this common childhood disease.

 XML Download
XML Download