

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Waldenstrom's macroglobulinemia (WM) is a low-grade lymphoplasmacytoid malignancy that affects B lymphocytes. It is characterized by the excess production of immunoglobulin M (IgM) monoclonal (M) protein, which gives the name “macroglobulinemia.” Common presenting symptoms include pallor, oronasal bleeding, weakness, fatigue, weight loss, fever, and night sweats. Various systemic and neurologic symptoms, such as impaired vision, headache, stroke, and dementia, may also be seen due to hyperviscosity syndrome1. Many dermatologic problems have been reported in association with M proteins2, specifically in WM and other monoclonal gammopathies. Here we report a case of a 64-year-old man presenting with intensely pruritic papules and plaques associated with WM.
CASE REPORT
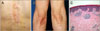
A 64-year-old man with no specific medical comorbidities presented with a 10-day history of severely itchy, erythematous to brownish papules and plaques on the trunk and both elbows (Fig. 1). The clinical features were suspicious for eczematous dermatitis, such as allergic contact dermatitis. Therefore routine laboratory studies and a patch test were performed, but no specific findings were noted, except for mild anemia as hemoglobin (Hb) 12.6 g/dl. Even total serum IgE levels were normal. He was treated with oral antihistamines and topical steroids, but the effects were minimal. Various treatments including narrow band ultraviolet B light therapy and immunomodulators such as dapsone and cyclosporine were also administered, but the severity of the pruritic skin lesions waxed and waned while progressively worsening. Only oral corticosteroids were effective at slightly improving the pruritus. Serum Hb decreased from 12.6 g/dl to 10.6 g/dl during the treatment over 6 months, so dapsone and cyclosporine were discontinued. The anemia persisted despite discontinuation, so the patient was referred to the Department of Hematology. Ferrous sulfate was administered for three months, but because the anemia did not improve, additional hematologic studies were performed. Lymphocytosis was shown on peripheral blood smear, the serum IgM level was elevated at 2,287 mg/dl, and serum electrophoresis revealed a monoclonal peak at the β1 region (M-protein 21%, 1.7 g/dl; Fig. 2A). Bone marrow biopsy revealed 80% cellularity showing lymphoid cell aggregation with CD20+, CD79a+ and CD5− (Fig. 2B~D). As such, the diagnosis of WM was established. This was one year and four months after the patient first visited our clinic for pruritic skin lesions. Because the distribution and morphology of skin lesions were similar to that of WM cutis, a 3-mm punch biopsy was taken to clarify this association. The histopathologic findings revealed hyperkeratosis, parakeratosis and patchy infiltration of mixed lymphohistiocytes (Fig. 1C). The immunohistochemical stain revealed no deposition of lambda or kappa chains, and the infiltrating cells were negative for CD79a. Thus, it is thought that the skin manifestations were probably not due to direct infiltration of tumor cells or deposition of M protein.
The hematologists recommended bone marrow transplantation, but the patient persistently refused. Over the course of 2 years, he was managed on cyclosporine, antihistamines, and topical corticosteroids, but he complained of worsening pruritus. Cyclosporine was the only effective drug for the pruritic lesions, but its effect was minimal. He recently started rituximab, cyclophosphamide, and dexamethasone (RCD) therapy. His anemia and absolute neutrophil count gradually improved, and the pruritus also subsided after five sessions of RCD. He was only taking loratadine for pruritus at the time of the first RCD session, and he did not require any antihistamines or topical corticosteroids after the fifth session.
DISCUSSION
WM is an indolent lymphoproliferative disorder affecting B lymphocytes. It is characterized by monoclonal IgM gammopathy and bone marrow infiltration with lymphoplasmacytoid cells3. The characteristic clinical features include anemia, lymphadenopathy, and hyperviscosity syndrome4. It is a relatively rare disease with an incidence of three per million people per year and the median age at diagnosis is 64 years. Patients with WM typically exhibit bone marrow with over 10% infiltration by clonal lymphoplasmacytic cells and a monoclonal IgM gammopathy in the blood. Various dermatologic conditions have been reported in association with WM and other monoclonal gammopathies such as multiple myeloma15. Dermatologic conditions with WM are rare6, appearing in only about 5% of cases. These lesions evolve due to various pathological processes, including direct infiltration of tumor cells into the skin, deposition of paraproteins, hyperviscosity syndrome and cryoglobulinemia of the blood (Table 1)47.
The cutaneous conditions caused by a monoclonal gammopathy can be classified into four groups8. The conditions in group I result from the extension and proliferation of malignant plasma cells into the skin or the cutaneous deposition of M proteins. Macroglobulinemia cutis which typically presents with pruritic papules and plaques of the trunk and extensor surfaces of the extremities9 is classified in this group. Group II is strongly associated with monoclonal gammopathy but is not the result of infiltrating malignant cells or deposition of M proteins in the skin. It may also be associated with paraproteinemia. Scleromyxedema, scleroderma, necrobiotic xanthogranuloma, plane xanthoma, and Schnizler syndrome are reported to be associated with this category. The dermatoses in group III are assumed to be associated with a monoclonal gammopathy, but the incidence of paraproteinemia has not been shown to be greater in these conditions than in other dermatoses. Autoimmune-blistering diseases, malignant skin conditions, and other various dermatoses are classified in this category. Subepidermal bullous dermatosis, xanthoma disseminatum, and non-Hodgkin's lymphoma are known to be associated with this category. The dermatologic manifestations in group IV result from impaired immunity and increased susceptibility to infection. These problems are caused by decreased functional immunoglobulin, kidney injury caused by M proteins, and hyperviscosity syndrome. Various conditions including purpura, nonspecific pruritus, and infection are classified in this category.
The pruritic papules and plaques of the trunk and extensor areas, as shown in this case, are the typical clinical features of macroglobulinemia cutis. However, the immunohistochemical stain for CD79a was negative, and neither lambda nor kappa chain deposition was identified in this case. The infiltrating cells were not thought to be WM tumor cells, and as such, the histologic findings were not diagnostic for macroglobulinemia cutis. Despite these negative findings, the severely pruritic papules and plaques were thought to be strongly associated with monoclonal gammopathy because they appeared at the time of WM diagnosis and progressively worsened with disease progression. The lesions were unresponsive to various conventional treatments; however, they improved with RCD therapy. These dermatologic features have not been previously documented as a nonspecific cutaneous manifestation of WM or monoclonal gammopathy.
The only reported case of WM presenting with pruritic papules on the trunk and upper limbs exhibited no histologic infiltration of tumor cells in the skin, and immunofluorescence showed plasma cells with polyclonal positivity for heavy chains, similar to our case10. The patient in the previously reported case also did not benefit from antihistamines, potent topical corticosteroids, chlorambucil or cyclophosphamide, and his pruritus was managed only partially by plasmapheresis.
The macroglobulinemia cutis-like manifestation of pruritic papules and plaques distributed on the trunk and extensor surfaces can be due to not only direct infiltration of malignant cells or deposition of M protein, but also due to paraproteinemia or unknown mechanisms. Considering that most diseases that present with pruritic papules and plaques are not accompanied by paraproteinemia, our case could pertain to group III of the monoclonal gammopathy-associated skin diseases. To clarify the association between intensely pruritic papules/plaques and Waldenstrom's macroglobulinemia, more reports and further studies could be needed.


 XML Download
XML Download