

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Acne is one of the most common inflammatory skin diseases with accumulated data regarding the pathogenesis of acne but the treatments are still unsatisfactory.
A related family of toll-like receptors (TLRs) is widely expressed in mammalian tissues, particularly epithelia, and a growing body of research indicates the importance of innate immunity as defence response in skin diseases1. The TLR family is composed of a group of cell surface receptors that are important for both innate and acquired immune responses against invading microorganisms2. Pathogen-associated molecular patterns bind to TLRs in order to induce various inflammatory reactions mediated by nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) pathways3.
Propionibacterium acnes, a commensal Gram-positive anaerobic rod is considered to be the primary bacterium implicated in acne development4. However, successful antibiotic treatment may not result from a reduction of bacterial numbers. The antibiotics typically used in the management of acne, tetracyclines, have additional anti-inflammatory actions independent of microbial killing. P. acnes has role in inciting inflammation in acne, thus more targeted therapeutic interventions may be necessary. P. acnes triggers inflammatory cytokine responses in acne by activation of TLR2 indicating that TLR2 could be a target for treatment of acne. For one example, oral isotretinoin treatment normalized exaggerated TLR2-mediated innate immune responses in acne patients. P. acnes also activates inflammatory molecules such as interleukin (IL)-1α, IL-8 and tumor necrosis factor (TNF)-α via the TLR2 signalling pathway5. The release of proinflammatory cytokines mediated through TLR2 has a harmful effect to acne as it promotes inflammation and tissue destruction6. Several reports have shown that TLRs communicate with the epidermal growth factor receptor (EGFR) via epithelial signalling to produce certain innate immune responses7. However, the effect of epidermal growth factor (EGF) and EGFR signalling on the TLR2 associated pathway in acne and other inflammatory skin diseases is unknown.
The EGFR signalling pathway has a critical role in skin development and homeostasis. Epidermal keratinocytes are a rich source of EGFR ligands including EGF, transforming growth factor alpha (TGF-α), amphiregulin, heparin-binding EGF (HB-EGF) and epiregulin8. It is well known that EGF is involved in cutaneous wound healing through the stimulation, proliferation and migration of keratinocytes, endothelial cells and fibroblasts, thus facilitating epidermal and dermal regeneration9. Moreover, the local administration of EGF induced remission in patients with ulcerative colitis10. Interestingly, EGF was also involved in down-modulation of inflammation. In their report, EGFR signalling blunted allergen-induced IL-6 production and T-helper 17 cell responses in the skin, and attenuated the development and relapse of atopic dermatitis in animal study.
Although the important roles of EGF in wound healing are well known11, there are few reports about the effects of EGF and EGFR on inflammatory skin diseases including acne and human defence response. Considering that topical EGF preparation often has been used to improve acne in dermatological clinical setting and cosmetics including enough concentration of recombinant human EGF (rhEGF) improved the clinical severity of mild to moderate acne in one report12. Moreover, EGF receptor inhibitors which are often used to treat various cancers inevitably induced acneiform eruption in the skin as adverse effects of the drugs.
Thus, we investigated whether administration of rhEGF can affect P. acnes-induced events in normal human epidermal keratinocytes (NHK). Also, we hypothesized that EGF might downregulate TLR2 signalling pathway associated proinflammatory responses induced by P. acnes.
MATERIALS AND METHODS
Reagents
P. acnes (KCTC 3314) was obtained from the Korean Collection for Type Cultures (KCTC, Daejeon, Korea). rhEGF (Daewoong Pharmaceutical Co., Ltd., Seoul, Korea) was used in this study. Nicotinamide was purchased from Sigma-Aldrich (St. Louis, MI, USA). Gefitinib was purchased from AstraZeneca Corporation (San Diego, CA, USA).
Cell culture
NHK (Thermo Fisher Scientific, Waltham, MA, USA) were cultured in EpiLife Medium (Thermo Fisher Scientific) with human keratinocyte growth supplement (HKGS, Thermo Fisher Scientific). Before reagent treatment, the cells were cultured in EpiLife medium (Thermo Fisher Scientific) for starvation overnight. Cells were maintained in a humidified atmosphere of 5% CO2 at 37℃, and the medium was replaced every two days. To induce inflammatory cytokines and TLR2 signalling, NHK were stimulated by 10 multiplicity of infection (MOI) P. acnes. The reagents were treated to NHK with 10 MOI P. acnes.
Real-time reverse-transcription–quantitative polymerase chain reaction
After treatment, the total ribonucleic acid (RNA) was extracted from cells using the ReliaPrep™ RNA Cell Miniprep System (Promega, Madison, WI, USA), and 1 µg of the total RNA was converted to cDNA using the TaKaRa RNA PCR Kit ver. 2.1 (TaKaRa Bio Inc., Shiga, Japan), under the following reaction conditions: 45℃ for 45 min and 95℃ for 5 min. Probes were obtained from Applied Biosystems (Foster City, CA, USA) as Assays-on-Demand™ Gene Expression Assays (glyceraldehyde-3-phosphate dehydrogenase [GAPDH]: Hs02758991_g1, IL-1α: Hs00174092_m1, IL-8: Hs00174103_m1, TNF-α: Hs01113624_m1, TLR2: Hs01872448_s1, NF-κB: Hs00 765730_m1, p38α: Hs01051152_m1). Reactions were carried out on the ABI StepOnePlus™ (Applied Biosystems), and relative transcription levels were determined by GAPDH as the reference gene. The data were analysed by using the ABI StepOnePlus™ software (Applied Biosystems).
Enzyme-linked immunosorbent assay
The secretion of IL-1α, IL-8 and TNF-α in the 48 hours supernatants of reagent-treated or untreated control was determined using specific enzyme-linked immunosorbent assay (ELISA) kits as instructed by the manufacturer. The IL-1α, IL-8 and TNF-α ELISA kits were obtained from R&D Systems (Minneapolis, MN, USA). After treatment, whole-cell lysates were collected by using a cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA). The concentration of TLR2 in the cell lysates was then measured by using a specific ELISA kit (R&D Systems).
NF-κB (p65) activation assay
To examine transcription factor activity, nuclear extracts were collected using a nuclear extraction kit (Cayman, Ann Arbor, MI, USA) in the reagent-treated or untreated NHK for 48 hr. Protein concentrations were determined with a Bio-Rad reagent (Bio-Rad Laboratories, Hercules, CA, USA). The activation of NF-κB (p65) transcription factor was detected by using an NF-κB (p65) transcription factor assay kit (Cayman). Briefly, 10 µg of nuclear proteins were added to the wells with a complete transcription factor buffer and incubated overnight at 4℃. Blank wells, a positive control, and nonspecific binding samples were also included on the plate. After that, NF-κB binding was detected by incubating with monoclonal and secondary antibodies directed against the NF-κB p65 subunit. Reaction was quantified at 450 nm. The percent change in the activity of each test sample relative to the average of untreated samples was determined.
Statistical analysis
All experiments were carried out in triplicate, and the results are exposed as mean±standard deviation. p-values <0.05 were considered statistically significant. One-way analysis of variance with Dunnett's posttest was performed using GraphPad Prism ver. 7 (GraphPad Software, San Diego, CA, USA).
RESULTS
Downregulation of rhEGF on the expression of cytokines induced by P. ances
To investigate our hypothesis, we treated NHK with 10 MOI P. acnes and various concentrations of rhEGF for 6 and 48 hours. After that, we confirmed the mRNA and protein expressions of proinflammatory cytokines such as IL-1α, IL-8 and TNF-α through real-time quantitative polymerase chain reaction (RT-qPCR) and by using ELISA kits. The mRNA expression of IL-1α, IL-8 and TNF-α was induced by P. acnes and was downregulated by rhEGF in a concentration-dependent manner (Fig. 1A). Moreover, in a concentration-dependent manner, rhEGF inhibited the protein expression of IL-1α, IL-8 and TNF-α increased by P. acnes (Fig. 1B). Nicotinamide of 5 µM, which was used as a positive control, reduced the mRNA and protein expressions of proinflammatory cytokines including IL-1α, IL-8, and TNF-α (Fig. 1).
Regulation of rhEGF on the TLR2 expression in NHK stimulated by P. ances
Next, we assessed the TLR2 gene and protein expression in NHK when P. acnes and various concentrations of rhEGF were added to the culture for 6 and 48 hours through RT-qPCR and by using ELISA kits. As expected, 1 and 10 ng/ml rhEGF as well as 5 µM nicotinamide inhibited the TLR2 gene expression in NHK stimulated by P. acnes (Fig. 2A). Moreover, the protein expression of TLR2 increased by P. acnes was inhibited by rhEGF and nicotinamide (Fig. 2B).
Regulation of rhEGF on the NF-κB activity in P. acnes-treated NHK
Then, to determine which transcription factors are involved in these results, we confirmed the mRNA expression of NF-κB and P38α. NF-κB expression, but not P38α RNA expression, was downregulated by 1 and 10 ng/ml of rhEGF (Fig. 2C, D). On the other hand, P38α RNA expression, but not NF-κB expression, was downregulated by 5 µM nicotinamide in P. acnes-treated NHK (Fig. 2C, D). Similarly, with gene expression, the resulting NF-κB activity was also inhibited by rhEGF in a concentration-dependent manner in P. acnes-treated NHK (Fig. 2E).
Attenuation of EGF's regulatory effects on the proinflammatory cytokines, TLR2 expression and NF-κB activity induced by P. acnes
To confirm the inhibitory effects of rhEGF on proinflammatory cytokines and TLR2 expression, we used gefitinib, which is one of the EGFR inhibitors. NHK were treated with 100 nM of gefitinib overnight. Then, they were treated with 10 MOI P. acnes as well as 1 and 10 ng/ml rhEGF for 48 hours. As shown in Fig. 3, the addition of gefitinib reversed the inhibitory effects of rhEGF on the protein expression of IL-1α, IL-8 and TNF-α. Next, to investigate the effects of gefitinib on TLR2 expression, we estimated the TLR2 gene and protein expression in NHK treated with gefitinib and/or 10 MOI P. acnes and/or rhEGF. Interestingly, the TLR2 protein expression, but not the mRNA expression of TLR2, inhibited by rhEGF was partially recovered by 100 nM gefitinib (Fig. 4A, B). Similarly, as shown in Fig. 2C, the mRNA expression levels of p38α were not altered by rhEGF and gefitinib (Fig. 4C). As expected, the mRNA expression of NF-κB and NF-κB activity downregulated by rhEGF were again recovered by gefitinib (Fig. 4D, E).
DISCUSSION
Treatments for acne vulgaris target one of more of the steps in pathogenesis and may be administered topically or orally. The most widely used acne treatments are topical formulations. Nicotinamide, also known as niacinamide, is the amide derivative of vitamin B3. It has been used both topically and systemically in various cutaneous inflammatory disorders including acne vulgaris. Recent studies revealed that nicotinamide decreases the in vitro secretion of IL-8 in P. acnes induced keratinocytes and has an anti-inflammatory role via inhibition of lysosomal enzyme release and mast cell degranulation13. In this study, nicotinamide was used as a positive control and was used to compare the inhibition effect of rhEGF on inflammatory cytokines expression. As the results, rhEGF inhibited mRNA and protein expression of IL-1α, IL-8 and TNF-α, similar to nicotinamide. These results suggest that EGF can also have potential role to be used as acne vulgaris treatment.
Although the role of EGF in the skin regeneration process, such as in wound healing, is well established, its role in innate immunity and inflammation is not well characterised. In this study, we demonstrated that EGF/EGFR signalling plays an important role in the regulation of inflammation and innate immune responses by inhibiting proinflammatory cytokines including IL-1α, IL-8 and TNF-α through the suppression of NF-κB and possible relation of TLR2-NF-κB signalling in P. acnes-stimulated epidermal keratinocytes. The role of EGFR signalling in inflammation is contradictory depending on the reports. In psoriatic lesions, IL-8 gene expression was actively induced by EGFR ligands such as TGF-α and amphiregulin14. However, it is also reported that EGF treatment increased anti-inflammatory cytokine IL-13 transcription expression15. Moreover, EGFR ligands, such as TGF-α, HB-EGF and amphiregulin downregulated the levels of CCL2/monocyte chemotactic protein-1, CCL5/regulated-on-activation normal T-cell expressed and chemoattraction CXC ligand 10/IP101617.
In the present study, we demonstrated for the first time the dose-dependent reduction of the gene expression of proinflammatory cytokines, including IL-1α, IL-8 and TNF-α, in rhEGF-treated NHK stimulated by P. acnes. In accordance with the gene expression data, the protein expression of cytokines measured using ELISA kits was markedly inhibited by rhEGF in P. acnes-induced epidermal keratinocytes. We also demonstrated that the expression of TLR2 and the activation of NF-κB were downregulated by rhEGF in P. acnes-treated NHK. From our results, we can suggest that EGF/EGFR signalling in bacteria-stimulated epidermal keratinocytes may help to reduce inflammatory responses by downregulating proinflammatory cytokines.
Large amounts of TLR2 have been found to be expressed on perifollicular and peribulbar macrophages in acne lesions18. In addition, it has been shown that there is a positive correlation between the severity of acne lesions and the concentration of cells expressing TLR218. TLR signalling can be divided into two signalling pathways: MyD88-dependent and MyD88-independent pathways219. It is known that TLR2 uses a Myeloid differentiation primary response gene 88 (MyD88)-dependent signalling pathway20. When TLR2 recognizes lipoprotein or lipopolysaccharide (LPS), the signalling is initiated. MyD88 recruits the interleukin-1 receptor-associated kinase 1 family of proteins, and by sub-signalling the p50 and p65 of NF-κB's subunits are translocated into the nucleus. The activated NF-κB in the nucleus initiate the transcription of inflammatory cytokine genes20. TLR2 can recognise P. acnes peptidoglycan and lipoteichoic acid, and it is considered that P. acnes-induced innate immune response through the TLR2 signalling pathway, including NF-κB activation in the cells of the pilosebaceous unit and infiltrating monocytes, contributes to acne pathogenesis21. NF-κB is a protein group that regulates inflammatory response, the immune system, cell death, cell proliferation, and the differentiation of epithelial cells22. It controls the expression of various genes and forms the central axis of the intracellular signal transduction system. In particular, NF-κB plays a role in the regulation of immune homeostasis and inflammation in the skin. Recent study revealed that insulin like growth factor-1 increased inflammatory biomarkers via activating NF-κB in cultured sebocytes23. According to An et al.24, pyrrolidinecarbodithioic acid, which is a potent NF-κB inhibitor, inhibited LPS-induced NF-κB p65 subunit nuclear translocation as well as the up-regulation of TLR2, 4 and 9 mRNA expression in dendritic cell. That results mean that the NF-κB pathway is important in the regulation of TLR expression24. In addition to NF-κB activation, TLR2 also activates MAPK signalling. In this signalling, p38 and c-Jun N-terminal kinase activation and finally Elk-1 and activator protein 1 are translocated into the nucleus2526. In this study, rhEGF inhibits the expression and activity of NF-κB induced by P. acnes but P38α expression was not regulated by P. acnes and rhEGF in NHK. This result suggests that the immune responses induced by P. acnes are mediated by TLR2 signalling, which activates NF-κB.
EGF binds to a homodimer or heterodimer receptor of EGFR to activate EGFR. Activated EGFR initiates underlying signalling pathways such as Ras/MAPK, Janus kinase/signal transducers and activators of transcription (JAK/STAT), and phosphatidylinositol 3-kinase/a serine/threonine kinase. The activated signalling pathway is involved in the transcription of various genes, including NF-kB27. Suppressor of cytokine signalling (SOCS) refers to a family of genes involved in inhibiting the JAK/STAT signalling pathway28. There are 8 members of the SOCS family; cytokine-inducible SRC homology 2 (SH2)-domain-containing protein and SOCS1-729. Recently, SOCS proteins have been involved in inflammatory responses of skin tissue. Expression level of SOCS3 protein is increased in peripheral blood mononuclear cells form patients with atopic dermatitis30, psoriatic lesional skin31. Interestingly, SOCS3 is induced during acute wound inflammation and remarkably, EGF induces SOCS3 MRNA expression in keratinocytes and murine primary keratinocytes32. Inferred from the references, it is speculated that SOCS proteins may be involved in these results that rhEGF inhibited cytokines expression and inhibition of NF-κB activity. The further study is needed on the relation between inhibition effects of EGF on inflammatory responses and SOCS proteins.
In tumor cells, EGF activates NF-κB related signalling pathways, and these signalling cascades are believed to crucially contribute to tumor development and progression33. Thus, EGFR inhibitors, such as erlotinib and gefitinib, are being used as therapeutic agents for various types of cancer34. However, cutaneous side effects are often observed in patients treated with EGFR inhibitors35. They include hair loss, acneiform eruption, paronychia and xerosis. EGFR inhibition enhances the production and release of proinflammatory mediators, and inhibits the production of antimicrobial peptides, resulting in the development of inflammatory papules and pustules as well as an impaired host defence function3637. Interestingly, in this study, rhEGF also inhibits the expression of TLR2 and proinflammatory cytokines increased by P. acnes through the inhibition of NF-κB, which is restored by using gefitinib.
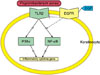
Our results indicate that EGF has an important role in the regulation of skin inflammation and immune responses through the suppression of NF-κB activity in relation with TLR2/ NF-κB signalling (Fig. 5). These findings, therefore, suggest the possible benefits of EGF in the treatment of acne and other inflammatory skin diseases.




 XML Download
XML Download