

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Dravet syndrome (DS) is one of the most intractable epileptic encephalopathies, and it is characterized by febrile seizures beginning in the first year of life, multiple seizure types, and psychomotor function regression.12 About 70% of DS patients are known to carry mutations in the sodium channel alpha-1 subunit gene (SCN1A).345 It has been revealed that malfunction of inhibitory neurons underlies the brain hyperexcitability evident in epilepsy patients with SCN1A mutations.6
Stiripentol (STP) at clinically relevant concentrations enhances central GABA neurotransmission by increasing the duration of GABA-A receptor channel opening and interferes with GABA reuptake and metabolism.789 STP was approved for DS as an orphan drug throughout Europe and other countries in the early 2000s, and it is still the only drug specifically indicated for DS in combination with valproate and clobazam.10 Previous studies have found that this combination shows short-term efficacy in reducing the frequency and duration of seizures.
However, the etiology of DS remains unknown in about 20% of DS patients, and additional genes including GABRG2 and SCN1B are probably implicated.11 In the present study, we therefore aimed to determine the efficacy of STP in controlling seizures according to the presence of definite SCN1A mutations in patients with DS.
METHODS
Patients and stiripentol efficacy assessments
In total, 32 unrelated pediatric patients were clinically diagnosed with typical DS at Severance Children's Hospital from January 2007 to May 2015. All of these patients met the following criteria:12 1) prolonged febrile and nonfebrile seizures within the first year of a life, 2) many different seizure types including myoclonic seizures, 3) frequent seizures when ill or having a fever during childhood, and 4) normal development in the early years followed by progressive developmental delay.
In accordance with known guidelines,910 STP was introduced at a dosage of 25 mg/kg/day, with an ultimate target dosage of 100 mg/kg/day over 4 weeks. When a side effect occurred, the STP dosage was increased more slowly or adjusted in some other way so as to minimize the side effect. The occurrence of any drug interactions resulted in the maximum dosages of valproate and clobazam being decreased to 20 and 0.5 mg/kg/day, respectively.
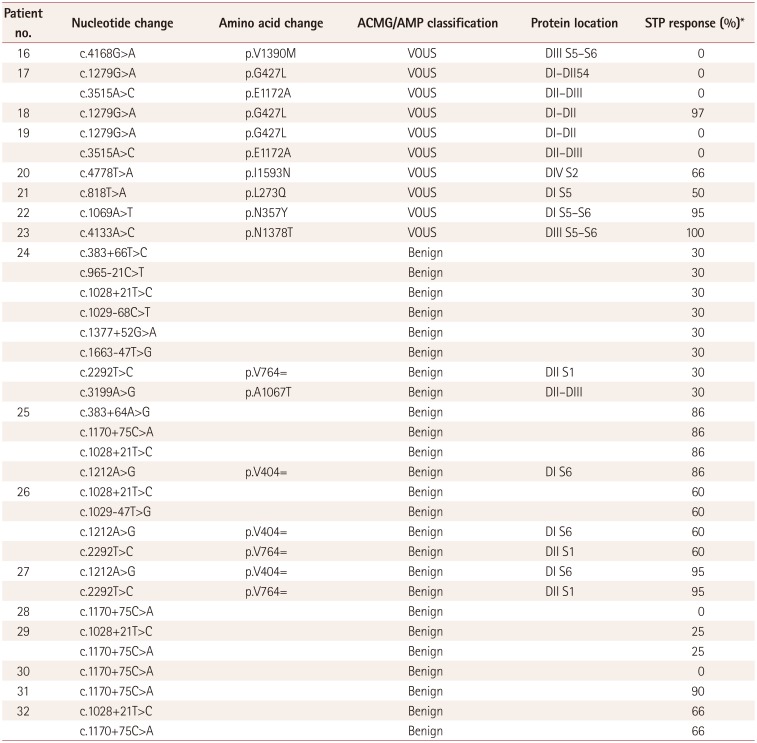
Thirty-two patients were classified into a mutation group with pathogenic/likely pathogenic variants (n=15) and a nonmutation group with variants of unknown significance (VOUS) or benign variants (n=17). All patients were followed up on a monthly basis, with information obtained using medical records and seizure diaries, and missing data collected by telephone calls. The baseline seizure frequency was obtained from the total number of all clinical seizures that occurred during the 12-week period preceding STP medication. The efficacy was assessed by comparing the baseline seizure frequency with that observed during the final 12 weeks before the last visit. The efficacy of STP in seizure control was compared between the two groups using the mean percentage reduction from the baseline seizure frequency as an exploratory efficacy variable. We also examined the efficacy of STP in patients according to the type of mutation (truncation or missense) and the protein location of the mutation.
In statistical analyses, independent t-tests were used to compare data between the mutation group (pathogenic and likely pathogenic) and the nonmutation group (VOUS and benign). Two-sided Fisher exact tests and Pearson's chi-square tests were also applied. PASW statistics software (version 18.0, SPSS Inc., Chicago, IL, USA) was used to implement the statistical analyses, and p values <0.05 were considered statistically significant.
SCN1A analysis and interpretation of variants
Direct sequencing of all coding exons and flanking intron sequences of SCN1A was performed using primer pairs designed by the authors. Sequence variations were analyzed by comparison with the corresponding wild-type sequence. When pathogenic or likely pathogenic variants were consistent with the patient's phenotype, final validation using another type of confirmatory assay and a parental study was planned. The variants were interpreted in accordance with the five-tier classification system recommended by the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) using a step-by-step approach. VOUS (and especially missense variants) were prioritized according to population frequency, ACMG/AMP score, and the patient's clinical phenotype.12 Parental studies were scheduled to detect de novo occurrence. For patients without mutations, the presence of copynumber variations including large exon deletions and duplications was confirmed by multiplex ligation-dependent probe amplification (MLPA) using the SALSA MLPA P245 Microdeletion Syndrome kit for SCN1A (MRC Holland, Amsterdam, the Netherlands).
The study protocol was approved by the Institutional Review Board of Yonsei National University Hospital (IRB No. 4-2016-0921). Written informed consent was obtained from the parents of all enrolled patients.
RESULTS
Patient demographics
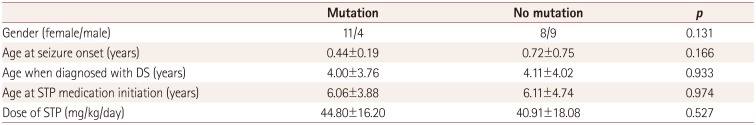
The 32 patients enrolled in this study comprised 13 males and 19 females with an age at seizure onset of 7.08±6.84 months (mean±SD; range 3.0–15.0 months). Their age at the initiation of STP medication was 73.08±51.48 months (range 12.0–192.0 months), their STP dosage was 42.73±17.06 mg/kg/day, and their medication duration was 34.44±144.0 months (range 3.0–81.0 months). We also compared the baseline characteristics between the mutation group (pathogenic or likely pathogenic variants) and nonmutation group. The STP dose did not differ significantly between the mutation group (44.80±16.20 mg/kg/day) and nonmutation group (40.91±18.08 mg/kg/day) (p=0.527), nor did any of the other clinical characteristics. The clinical and demographic profiles of the patients are summarized in Table 1.
SCN1A mutations
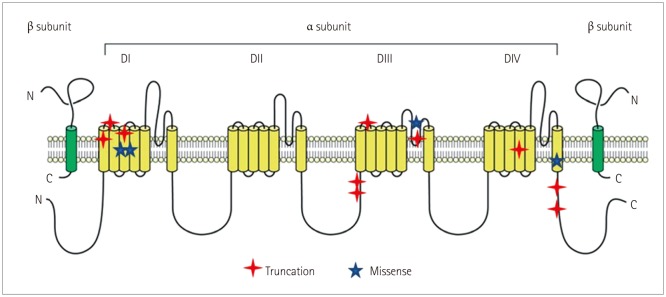
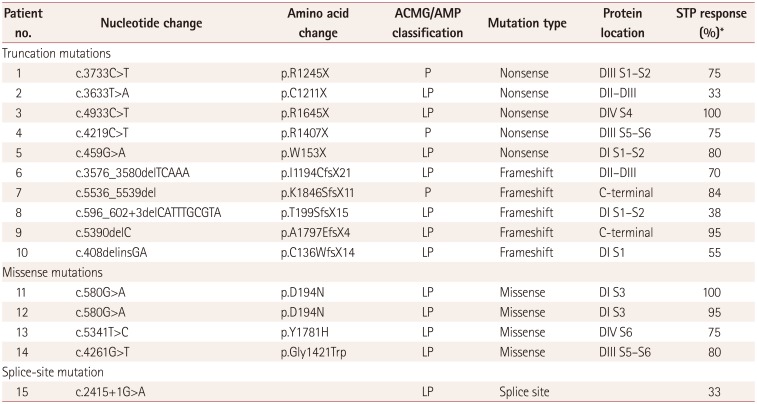
SCN1A analysis revealed pathogenic or likely pathogenic variants in 15 patients, comprising 10 truncations (5 nonsense mutations and 5 frameshift mutations), 4 missense mutations, and 1 splice-site mutation. The detailed genotypes are presented in Table 2. VOUS were present in eight patients, while nine patients had benign variants (Table 3). The locations of pathogenic and likely pathogenic variants were defined using a website (http://www.uniprot.org) showing gene locations (Table 2 and Fig. 1). Mutations were present at S1–S2 loop segments in three patients, at S5–S6 pore-loop segments in two patients, at linkers between domains in two patients, at the C-terminal in two patients, at the fourth transmembrane segment (S4) in two patients, and at other transmembrane segments in four patients.
Response to stiripentol according to SCN1A mutations
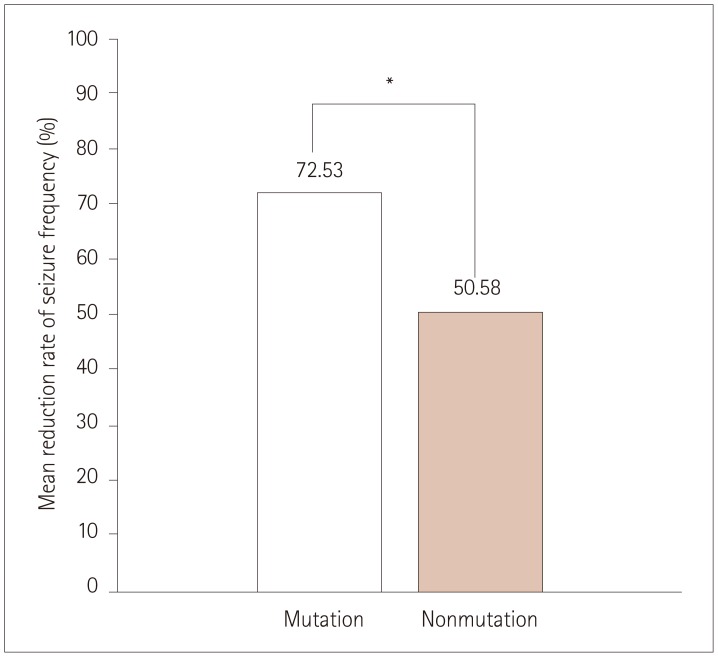
The seizure frequency reduced by 72.53±23.00% in the mutation group versus 50.58±40.14% in the nonmutation group (p=0.004) (Fig. 2). The efficacy of STP could unfortunately not be analyzed statistically according to mutation type or site in DS patients with mutations due to the small number of patients in each group (Table 2). Nonetheless, we did find the following characteristic tendencies: The seizure frequency reduced by 70.50±22.32% in 10 patients with truncation mutations and by 87.50±11.90% in 4 patients with missense mutations. In addition, according to the mutation site, STP was intriguingly 100% effective for two mutations in the voltage sensor segment (S4) and another segment (S3), while it did not exhibit favorable efficacy in three patients with mutations located at linkers between domains (DII–DIII), linkers between segments of domain I (DI S1–S2), or splice sites. Moreover, the efficacy of STP was similar regardless of the mutation site of SCN1A.
With regard to side effects of STP, two patients reported feeling sedated even after the STP dosages had been adjusted, and one patient experienced loss of appetite. No major side effects or adverse events were observed.
Discussion
We have compared the efficacy of STP between patients with pathogenic or likely pathogenic variants of SCN1A and those with VOUS or benign variants of SCN1A. STP combined with valproate and clobazam significantly reduced the seizure frequency in patients with DS due to definite SCN1A mutations.
DS provokes a devastating epileptic encephalopathy, and affected patients present with cognitive impairment, autistic behavior, and psychiatric disorders.12
De novo heterozygous mutations in SCN1A, which encodes a subunit of the neuronal voltage-gated sodium channel Nav1.1, result in faulty sodium channels in more than 70% of DS patients. SCN1A is a widely investigated gene due to its involvement in epilepsy and other neuronal disorders, and its expression in specific types of neurons and its role in the generation of action potentials have been studied intensively. The effects of SCN1A mutations on epilepsy have been revealed using SCN1A-specific knock-out or knock-in mice and a DS-specific reprogramming stem cell model, with the results indicating that the malfunction of inhibitory neurons is related to brain hyperexcitability.6
Based on the pathogenesis of DS, certain antiepileptic drugs (AEDs) including carbamazepine, oxcarbazepine, lamotrigine, and phenytoin should be avoided since they can aggravate seizures.1314 Conversely, drugs that enhance GABAergic neuron function including valproate, clobazam, and STP hold promise in controlling intractable seizures in patients with DS. Combining STP with valproate and clobazam may therefore be particularly effective in reducing the seizure frequency.
STP is a new-generation AED that enhances GABAergic transmission, and was first used to treat DS in the early 2000s.14 STP augments GABAergic activity by extending the mean open time of GABA-A receptors and increases GABA levels by interfering with its reuptake by acting as a positive allosteric modulator of these receptors. STP also inhibits lactate dehydrogenase, an enzyme that increases neuron activity by stimulating ATP-sensitive potassium channels. In addition, STP increases the concentration and duration of an AED by inhibiting cytochrome P450 enzymes.789 Many previous studies found that the seizure frequency was significantly lower for STP than in placebos.141516171819202122 Inoue et al.151617 conducted an open-label multicenter study and found that STP was an effective add-on therapy to reduce the seizure frequency in Japanese patients with DS. Chiron et al.18 conducted a randomized, double-blinded, placebo-controlled trial of add-on STP therapy in 41 patients with DS, and found 71% responders in the STP group compared to 5% responders in the placebo group. De Liso et al.22 investigated the efficacy of the STP in a cross-sectional study, and found that STP add-on therapy reduced the seizure frequency and severity in DS. However, none of these studies classified the effect of STP according to SCN1A mutation, since all patients with DS were included irrespective of whether or not they had a mutation. In contrast, the present study compared the efficacy of STP in the presence or absence of SCN1A mutations in patients with DS, with the results showing that STP was more effective in patients with definite SCN1A mutations.
SCN1A consists of four domains (DI–DIV), six transmembrane segments (S1–S6) including voltage sensor (S4) and pore-loop (S5–S6) regions, and connecting cytoplasmic loops or linkers.45 Ishii et al.23 reported that truncation mutations were distributed throughout the SCN1A protein in Japanese DS patients with mutations affecting particular regions within the channel, with half of missense mutations being in the pore region. We also demonstrated that truncation mutations are distributed more randomly in specific areas of the sodium channel. Ishii et al.23 also examined the response to AEDs as an add-on therapy according to the SCN1A mutation type (truncation or missense variants), and found that the most effective AEDs in patients with truncation mutations were STP, topiramate, and bromide (in decreasing order of efficacy), whereas the most effective AEDs in patients carrying missense mutations were clonazepam, bromide, and topiramate (also in decreasing order of efficacy). In contrast to a previous report,23 we found that efficacy of STP was somewhat better in patients with missense mutations than in those with truncation mutations; however, the small number of patients with missense mutations meant that statistical analysis could unfortunately not be performed. Nonetheless, we can suggest that STP is effective even in missense mutations if these mutations cause changes in the structure of sodium channels that contribute to pathogenesis. We also investigated whether STP was more effective in patients with mutations at specific locations, but failed to find any significant differences. One particularly notable finding was that seizures were poorly controlled in patients with SCN1A mutations at linkers between segments or domains other than in the pore-forming region. These results might be related to pathomechanisms of inhibitory neurons, but further research is required to elucidate the underlying electrophysiologic mechanisms in detail.
This study was subject to some limitations, including the relative smallness of the sample and it being designed as a retrospective review of clinical data. Nonetheless, we were able to demonstrate that STP has better efficacy in DS patients with definite SCN1A mutations than in DS patients with VOUS and benign SCN1A mutations. Further comparative prospective studies with larger samples are needed to determine the efficacy of STP according to the presence of SCN1A mutations.
 XML Download
XML Download