

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Carcinoid tumors are rare and mainly occurs in the gastrointestinal system (90%)1 originating from the neuroendocrine cells. The liver is only occasionally involved as a primary tumor site (0.59% of gastro-entero-pancreatic carcinoid tumors), but, on the other hand, the liver is the most common site of metastasis.234 The histological features of neuroendocrine tumors (NETs) depends on anatomical site and cell origin, however, they share relevant features such as macroscopic and microscopic characteristics.5 Primary liver NETs (PLNETs) share some common histopathological and clinical features with gastrointestinal NETs. They usually rise slowly,1 and the most frequently produced hormones are gastrin and chromogranin A.6 However, a clinical presentation with carcinoid syndrome is relatively uncommon in PNETs. The most frequently reported symptom is abdominal pain (33%), followed by the absence of symptoms (23.2%).6 The 5-year recurrence rate has been reported to be between 26–45.5%.7 There exist only a few reports on the treatment of recurrent PLNETs (rPLNETs). We report a rare case of long-term recurrence of PLNET after extended right hepatectomy in a 52-year-old woman.
CASE
In 2010, the World Health Organization (WHO) updated NET grading, mostly based on the Ki67 index and mitotic count, which is highly relevant about prognosis.8 In order to consider data in accordance with last WHO update on NET grading, we reviewed the literature since 2010 to until date based on the following exclusion criteria: lack of data about diagnosis and liver transplantation. We searched Pub med, Embase, Web of Science and Cochrane online databases with the following keywords: (neuroendocrine OR net OR carcinoid) AND (liver[title] OR hepatic[title]) AND primary[title], and obtained 303 results. Two investigators extracted the data independently (GP, CC). To avoid systematic biases, the authors independently reviewed all the eligible studies until a complete concordance was reached for all the assessed variables. Disagreements were resolved by discussion; with the participation of a third author (AR). Extracted data included demographic data, patient's characteristics, methodological data, overall survival (OS), disease-free survival (DFS) hazard ratio (HR), progression-free survival (PFS) HR, and postoperative complications.
Case report
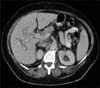
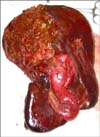
A 52-year-old woman with recurrent abdominal pain clinically compatible with symptoms of pancreatitis was admitted to the Emergency Department in October 2007. Clinical examination revealed a palpable mass in the right upper quadrant. Blood sample examination revealed amylase 600 IU/L, gamma-glutamyl transpeptidase 372 IU/L; alkaline phosphatase 1309 IU/L, and total bilirubin 1.90 mg/dL. Computed tomography (CT) and magnetic resonance imaging (MRI) studies revealed a liver mass in hepatic IV and V segments, without the involvement of vessels. The imaging also revealed an endobiliary thrombus (Fig. 1). The CT scan did not reveal any other site of disease. Tests for hepatitis B and C were negative. Tumor markers were negative. An intrahepatic cholangiocarcinoma was supposed, then a right hepatectomy extended to the segment IVB and lymphadenectomy extended to the celiac axis was performed (Fig. 2).
The histological examination revealed a grey-white to the yellow mass measuring 6×5 cm, free of disease surgical margins. Thirteen disease free lymph nodes were removed during lymphadenectomy.
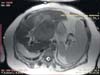
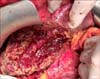
The immunohistochemical profile revealed that the carcinoma was Gramelius positive and the expression of epithelial markers such as CK, CK7, CK19 +/−, endocrine markers such as NSE (1+), chromogranin A (1+), and Ki67 (5%) were positive; while C-KIT, estrogen receptor, progesterone receptor, alfa-fetoprotein, chorioembryonic antigen (CEA), vimentin, and synaptophysin were negative. According to WHO 2010 classification, the tumor was a grade 2 PLNET. During a five year follow-up period, the patient did not show clinical or imaging features of recurrence. In April 2017, a remnant liver ultrasound examination revealed the presence of a liver mass. CT and MRI scans showed a remnant liver neoplasm measuring 8 cm (Fig. 3). After multidisciplinary discussion, repeat liver resection was performed (Fig. 4). The histological examination confirmed the diagnosis of recurrent PLNET. The patient was discharged 9 days after the resection and is alive and doing well.
Literature review
We found only 5 studies reporting about rPLNETs: 4 single case reports and 1 series, with 11 cases, globally, within the last 10 years. Ten cases followed our inclusion criteria.910111213
The patients' characteristics are presented in Table 1. The symptom at the time of presentation was abdominal pain in 4 cases, 4 patients had no symptoms, and 2 patients had diarrhea. Nine studies provided data about OS and DFS. Only seven studies reported tumor grading, and the results were quite heterogeneous within the reported cases. Median OS and DFS were 22 and 5 months, respectively. The main treatment modality for recurrent disease was transarterial chemoembolization (TACE), which was used in 4 cases, one patient was treated with chemotherapy, and in the remnant cases, there was no report about treatment modality. Only one case showed extrahepatic disease.
DISCUSSION
PLNET is considered a very rare disease. The true incidence is still debated because of the difficulty in differentiating the primary tumor from the metastatic NET. In fact, studies on gastrointestinal NETs reported a metastatic rate ranging from 32% to 75%, demonstrating liver as the main metastatic site.1415 And many cases considered PLNET, especially in the past, were patient with metastatic disease from occult gastrointestinal NET, as consequence of this, the main diagnostic issues about PLNET are related to its rareness, and the relatively high rate of liver metastasis from extrahepatic NETs. Consequently, the differentiation between PLNETs and extrahepatic NETS is difficult, even through histological examination.16 Accurate imaging, upper and lower gastrointestinal endoscopy and adequate follow-up are required in order to identify possible extrahepatic NETs. The largest review in English literature on this disease, reports about 150 cases.17
On the other hand, only 10% of PLNETs develop metastasis with a higher recurrence rate. This difference could be the consequence of biological features which are uncommon in other NETs.7 Some studies proposed that PLNET could originate from neuroendocrine cells of the intrahepatic biliary ducts, or from heterotopic cells located in the liver and that malignant cells could disseminate through the liver, determining the multifocal origin of many of these tumors.1819 According to these results, in our review, all the included patients were surgically treated with free of disease margins, 7 of 10 cases were multifocal, and only one case demonstrated extrahepatic metastases.
In all the included patients, recurrence occurred within, at most, 4 years after surgical treatment, whereas in our case report recurrence occurred after 9 years. To the best of our knowledge, our case report is the second reported case of PLNET long-term recurrence. We found another case of recurrence 13 years after resection.20 Another paper reported a case of long-term PLNET recurrence after resection, but in this case, the tumor already had metastatic localizations at the time of diagnosis.21
In all the included cases, preferred treatment was non-surgical, mainly using TACE (Table 1). However, when technically feasible, surgical treatment is considered as the most appropriate treatment, both for curative purpose and for cytoreduction in case of symptomatic malignancies. TACE is the preferred treatment in case of unresectable PLNET confined to the liver.17 Transplantation could be considered in highly selected patients.16
In our case, the patient was admitted in 2007 because of jaundice and pancreatitis. The diagnostic workup included a CT scan and the radiologic appearance was consistent with intrahepatic cholangiocarcinoma or a metastatic disease. Both colonoscopy and gastroscopy were performed in order to exclude the existence of other primary tumors. A biopsy was not considered and based on the radiological finding, a surgical resection was planned. Both jaundice and pancreatitis were secondary to the compression of the bile duct and the presence of a biliary thrombosis up to the bifurcation of the bile ducts that were removed from the stump of the right hepatic duct during surgery. The bile duct was drained through a trans-cystic drainage that was removed 30 days after surgery. After the histological results, the patient was followed up yearly, until a second neoplasm was detected in the ultrasound examination 9 years after the first operation. Some technical issues were discussed before the surgery for the recurrent neoplasm. The main portal branch of the remnant liver was in close relationship with the tumor, and a non-anatomical resection with R0 margin on the portal branch was considered feasible. Considering the good long-term outcome after the previous operation and despite the close relationship of the tumor with the main vascular and biliary structures, this surgical approach was considered acceptable. Only a small part of healthy parenchyma was sacrificed. TACE was also considered, but, due to the hypovascular pattern revealed in the imaging, we excluded this approach. Neoadjuvant TACE was discarded for the same reason. Moreover, in case of ineffective TACE, it is hypothesized that the progression of the disease and infiltration of the left portal vein or bile duct would definitively hamper the surgical approach.
In conclusion, PLNETs share some common features with NETs of another origin, such as slow rise, histopathological characteristics, and immunohistochemical patterns. On the other hand, these tumors differ based on the high rate of asymptomatic or paucisymptomatic presentation, the low incidence of carcinoid syndrome, and the relatively high rate of recurrence if compared with extrahepatic NETs. Finally, a longer follow-up period should be considered in order to avoid long-term recurrence.

 XML Download
XML Download