

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Developmental delay (DD) or intellectual disability (ID) occurs in 1–3% of the general population [1]. Even with recent advances in genetic testing, there are still patients without an etiologic diagnosis, who are on a long diagnostic odyssey to search for accurate etiology in some instances. This journey entails a heavy burden in medical costs as well as psychosocial stress on their families. One way in which the burden on patients, their families, and countries can be lightened is diagnosing these patients early and predicting possible complications, the prognosis, or the disease course; this allows them to receive appropriate clinical care [23]. As chromosomal abnormality is the main cause of DD/ID, conventional karyotyping is routinely used as a cytogenetic test; however, it has limitations of low resolution and a detection rate of only 3–5% [1]. There are still many unexplained cases of DD/ID in patients who have apparently normal karyotypes [14].
Chromosomal microarray (CMA) testing has been recommended as a first-tier cytogenetic diagnostic test for patients with DD/ID, autism spectrum disorders, or multiple congenital anomalies [4]. Since 2003, multiple reports have shown its importance in clinical practice [34]. CMA has an average diagnostic yield of 12.2% and can detect chromosomal aberrations of less than 5 Mb [4]. Although it has limitations in view of the interpretation of the pathogenicity of copy number variations (CNVs) [5] and in detecting balanced rearrangements or low-level mosaicism [4], it has the strengths of high detection rate and cost-effectiveness [6].
Even with its high diagnostic yield and clinical impact on pediatric care [23], CMA testing is not yet widely used for clinical diagnostic purposes in children with DD/ID in some countries, including Korea [378910]. We demonstrate the diagnostic utility of CMA testing for DD/ID in the pediatric population, by assessing data from a large cohort of Korean patients with DD/ID.
METHODS
1. Patients
We included 649 unrelated patients with DD/ID treated at the Seoul National University Children's Hospital, Korea. The cohort included 345 male and 304 female patients aged from seven months to 25 years. Blood samples were obtained from all enrolled patients whose parents provided informed consent. The medical records of the patients were reviewed retrospectively. The study protocol was approved by the Seoul National University Hospital Institutional Review Board (IRB No. 1511-099-722).
2. Karyotyping
The blood samples were incubated at 37℃ in MEM (Gibco/Life Technologies, Carlsbad, CA, USA) containing phytohemagglutinin for 72 hours. Colcemid (0.2 µg/mL) was added, and the culture was incubated for a further 40 minutes. Then, the samples were centrifuged at 185 g for 7 minutes. The dividing cells were harvested and incubated for 20 minutes at 37℃ in 0.075M KCl. The cells were centrifuged again and fixed in 3:1 methanol-acetic acid. Spread slides were prepared from centrifuged cells, and Giemsa banding was used to produce a visible karyotype. At least 25 karyotypes at 550-band resolution were prepared and analyzed for each sample. The chromosomes were classified using ChIPS-Karyo (GenDix, Seoul, Korea), and chromosomal abnormalities were reported according to the recommendations of the International System for Human Cytogenetic Nomenclature 2013 and 2016 [1112].
3. DNA preparation
Genomic DNA was extracted from peripheral blood leukocytes, using a QIAamp DNA Blood Midi Kit (Qiagen, Valencia, CA, USA), according to the manufacturer's instructions. When possible, parental DNA was collected.
4. CMA platform
CMA testing was conducted using Agilent Human Genome oligonucleotide comparative genomic hybridization (CGH) microarrays 1×244K, 4×180K, or 8×60K (Agilent Technologies, Santa Clara, CA, USA) with 8.9 kb, 13 kb, or 41 kb overall median probe spacing, respectively. In 21 cases, CGH+SNP microarray 2×400K, CGH+SNP 4×180K, or prenatal+SNP 4×180K, were used, which had 7.2 kb, 25.3 kb, or 13.4 kb overall median probe spacing, respectively. Genomic DNA was labeled and hybridized to the array, according to the manufacturer's protocol for Oligonucleotide Array-Based CGH for Genomic DNA Analysis (version 6.2; Agilent Technologies). A DNA reference sample (male or female human genomic DNA; Promega, Madison, WI, USA) was used. The slide was scanned on a microarray scanner (G2565CA; Agilent Technologies). Data were extracted from a *.tif image using Agilent Feature Extraction software (version 10.7.3.1) and analyzed with Genomic Workbench software (version 7.0.4.0, Agilent Technologies). The local background was subtracted from the median intensities of the Cy3 and Cy5 channels. The log2 patient-to-reference ratio was calculated for each spot and normalized to the median of the ratios of all chromosomes. All CNVs were called and based on human assembly GRCh37 (hg19).
5. Validation studies
Depending on the results, FISH or quantitative (q) PCR was performed in patients for verification, and in their parents for the presence of inheritance to identify pathogenicity of CNV. qPCR was performed using a ABI Prism 7500 system (Applied Biosystems, Foster City, CA, USA) with a fluorescent SYBR Premix Ex Taq (TaKaRa). Specific primer sequences and qPCR conditions are available on request. Raw data were analyzed using the comparative delta delta (ΔΔ) threshold cycle number (Ct) method. Standard FISH analysis was performed on metaphase chromosomes using commercially available or customized BAC clones (specific information can be provided upon request). Metaphase chromosomes were collected from the blood samples of patients and their parents and were analyzed using a fluorescence microscope (Nikon, Eclipse 80i, NY, USA) equipped with a computerized chromosome analysis system, ChIPS-FISH (GenDix, Seoul, Korea). At least 20 metaphase cells were analyzed for genomic imbalances.
6. Interpretation of CNVs
CNVs were considered pathogenic when they (1) overlapped with a previously reported pathogenic CNV/region; (2) were novel, but de novo CNVs were confirmed by parental testing; (3) included a gene within the CNV interval that had a compelling function or encompassed a well-known disease-causing gene; and (4) were inherited from a similarly affected parent, with a thorough evaluation based on the guidelines of American College Medical Genetics and Genomics (ACMG) and Miller et al [45]. In contrast, if identified CNVs were present in healthy normal controls (Database of Genomic Variants, DGV, http://projects.tcag.ca/variation) or gene-poor regions, they were considered non-pathogenic. The remaining CNVs were considered to be of uncertain clinical significance (VUS; where V=“copy number variations”, US=“uncertain significance”) [5]. In the present study, likely pathogenic VUS according to the ACMG guidelines [5] were reported as pathogenic because the CNVs had been described in only a single or a few case reports; however, pathogenicity was supported by parental testing. When the CNVs contained genes with compelling functions in the interval, but it was uncertain whether they were dosage sensitive, they were reported as VUS (no sub-classification). These VUS included cases with CNVs seen in both the patient and a healthy parent in this study, of which the pathogenicity had never been validated with parental testing in previous studies.
Whenever possible, to validate the genomic imbalance, we performed FISH or qPCR in the affected patient and both parents. To confirm the pathogenicity of CNVs found in the patients and their clinical significance associated with a phenotype, the Database of Chromosomal Imbalance and Phenotype in Humans using Ensemble Resources (DECIPHER, http://www.sanger.ac.uk/PostGenomics/decipher/), ISCA (ClinGen), PubMed, UCSC Genome Browser (http://genome.ucsc.edu/), and OMIM were used.
RESULTS
1. Detection rate of pathogenic CNVs
In total, 110 patients (53 male and 57 female) were found to have pathogenic CNVs (16.9%) (data available on request). Among these, 102 had a normal karyotype, as determined by conventional G-band karyotyping. One patient (P50) had a structural abnormality on chromosome 8, although it was not definite. Seven patients were tested with CMA without prior chromosome analysis (data available on request). Validation testing was performed in 65 patients (59.1%) with FISH (63 cases) or qPCR (2 cases).
2. Characteristics of pathogenic CNVs
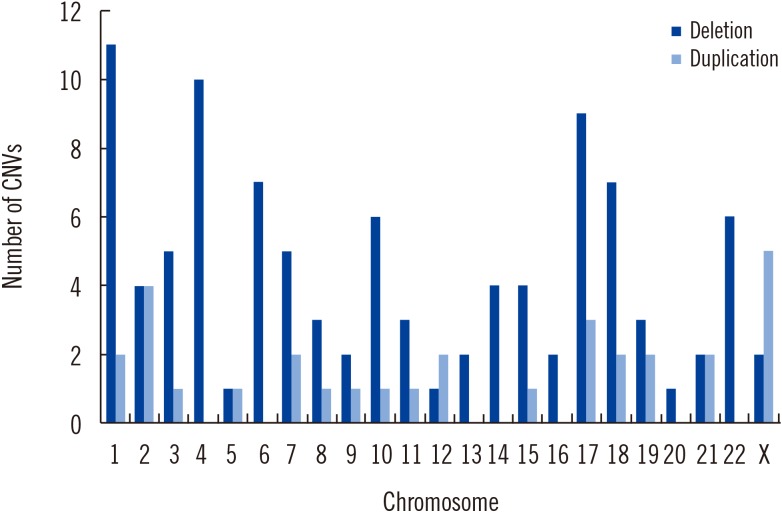
Pathogenic CNVs included 100 deletions and 31 duplications, ranging from 270 kb to 30 Mb. Each patient had 1.19 pathogenic CNVs on average. Most of the pathogenic CNVs (125/131, 95.4%) were larger than 500 kb, whereas six pathogenic CNVs smaller than 500 kb (6/131, 4.6%) were identified. They included one patient (P53) with a 320-kb 9q34.3 sub-telomeric deletion (Kleefstra syndrome), one patient (P109) with a 440-kb Xq28 duplication encompassing MECP2, one patient (P11) with a 440-kb 1q21.1 duplication, and one patient (P36) with a 450-kb 5q31.2 deletion encompassing PURA gene. Chromosomal distribution of the pathogenic CNVs is provided in Fig. 1.
Nineteen patients (19/110, 17.3%) had two or more pathogenic CNVs. Most of them had one deletion and one duplication, although there was one patient with two deletions, one with two deletions and one duplication, one with two duplications, and one with three deletions.
Parental testing was carried out for 66 patients (60.0%) by FISH (57 cases), qPCR (two cases), CMA (two cases), or karyotyping (five cases). Among them, 57 patients (86.4%) carried de novo pathogenic CNVs. Eight patients (12.1%) inherited CNVs from a healthy parent with a balanced translocation, in which case genetic counseling was provided. Among these, six patients (P6, P19, P30, P37, P46, and P89) had unbalanced translocations with both genomic loss and gain, whereas two patients (P18 and P95) had one duplication. The latter were inherited from their fathers with balanced translocations; the duplications were on 2q24 and 19p13, respectively. Additionally, a pathogenic CNV identified in one patient (P68) was inherited from a similarly affected mother with mild mental retardation and facial dysmorphism (See Supplemental Data Fig. S1).
3. Common pathogenic CNVs
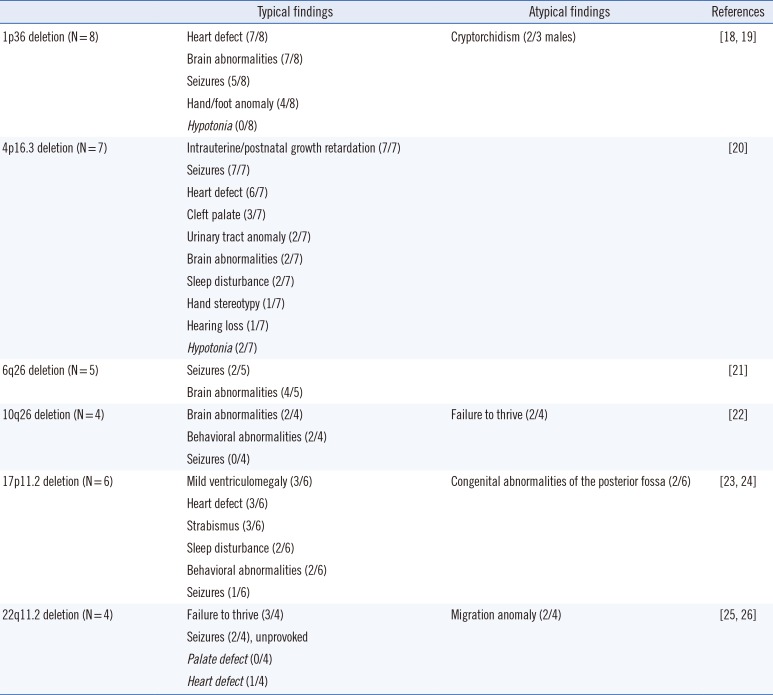
The most common pathogenic CNVs were 1p36 deletion (eight cases), 4p16.3 deletion (Wolf-Hirschhorn syndrome, seven cases), 17p11.2 deletion (Smith-Magenis syndrome, six cases), 6q26 deletion (five cases), 10q26 deletion (four cases), and 22q11.2 deletion (four cases). They had known CNV intervals, but did not always show all the typical features. As shown in Table 1, hypotonia, which is commonly involved in 1p36 deletion syndrome, was not observed in our patients, whereas cryptorchidism seen in a minority was involved in two out of three male patients with 1p36 deletion. Unexpectedly, congenital heart defects were commonly involved in patients with Wolf-Hirschhorn syndrome, and brain abnormalities, except ventriculomegaly, were seen in two out of six patients with Smith-Magenis syndrome. In addition, abnormal neuronal migration was involved in two out of four patients with DiGeorge syndrome, whereas cardiologic abnormalities, a characteristic of the syndrome, were observed in only one patient.
4. Identification of rarely reported pathogenic CNVs
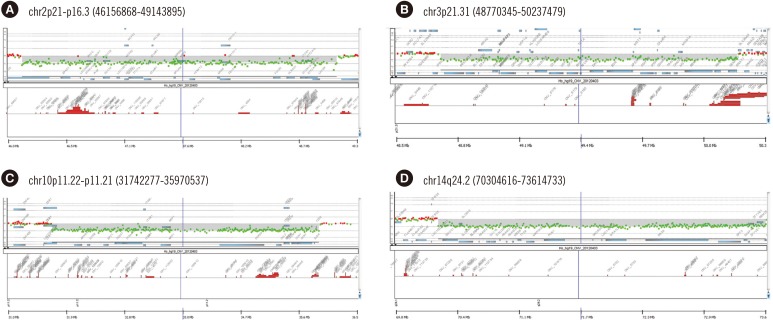
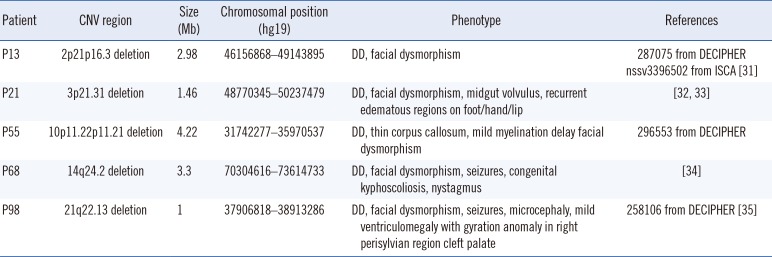
In five patients, we identified and confirmed rarely reported pathogenic genomic imbalances, which were considered as likely pathogenic VUS according to the ACMG guidelines [5] (Table 2 and Supplemental Data Fig. S2). They included regions that did not completely overlap with those of known genomic imbalances. Public databases revealed that in some of these regions, the pathogenicity remained uncertain. Five pathogenic CNVs that have been rarely reported included deletions on 2p21, 3p21.31, 10p11.22, 14q24.2, and 21q22.13. Available CMA images of the rarely reported pathogenic CNV regions are presented in Fig. 2. They were all verified with FISH (four cases) or qPCR (one case). One male patient (P68) had a 14q24.2 deletion inherited from his mother who was similarly affected, although she showed a mild phenotype.
DISCUSSION
Conventional karyotyping has been widely used to identify the causes of DD/ID in patients. Many cases with chromosomal aberrations are missed and remain without an etiologic diagnosis because conventional karyotyping cannot detect pathogenic genome imbalances of a size smaller than 5 Mb [1]. Furthermore, chromosomal abnormalities with a size larger than 5–10 Mb can be missed by G-banding [413]. In contrast, CMA can identify much smaller CNVs and is sensitive enough to identify pathogenic CNVs [1415], although its higher resolution may simultaneously result in a higher detection of non-pathogenic CNVs [141617]. There is also a limitation in the detection of balanced rearrangements, such as translocations and inversions or low-level mosaicism, but they account for less than 1% of chromosomal abnormalities [2]. Genome-wide CMA testing is a cost-effective method to establish an accurate diagnosis in patients with DD/ID.
We identified pathogenic genomic imbalances in 16.9% of children with DD/ID, even with the stringent criteria for pathogenicity used. We considered de novo status as evidence supporting pathogenicity by performing parental testing and validation studies in more than half of the patients. If we identified the same CNV in a healthy parent upon thorough examination, we considered it as a non-pathogenic CNV, although the possibility of incomplete penetrance or variable expressivity cannot be excluded. Pathogenic CNVs included 100 deletions and 31 duplications, with losses being approximately three times more common than gains [18]. The diagnostic yield in the present study was relatively high compared with those in previous studies on patients with DD [41619], although the yield can depend on patient selection criteria/number, prior evaluations, and the CMA platform used. In the present study, there were 16 patients with large CNVs (>10 Mb) that had been missed by karyotyping. Among them, seven patients had large rearrangements, which might have been missed using G-banded karyotyping because the translocations included one deletion and one duplication with similar band pattern or size. These findings indicate that CMA testing is preferable to conventional karyotyping, even in patients with large-sized CNVs [413]. Karyotyping can elucidate structural changes but is limited because of subjective assessment; it is therefore prone to inter-laboratory and inter-observer variation in detection [13]. CMA testing should be conducted as a first-tier test in patients with unexplained DD/ID, because chromosomal abnormalities are identified as genetic causes in a considerable portion of patients with DD/ID. Moreover, CMA testing of all children with unexplained DD/ID should be conducted, regardless of clinical severity [182021].
Taken together, three pathogenic CNVs accounted for 19.1% (21/110) of all causes of DD/ID in patients in the present study: 1p36 deletion, 4p16.3 deletion, and 17p11.2 deletion syndrome in that order. They were somewhat different from the recurrent syndromes commonly identified in other studies [1516]. This might result from the fact that the cohort in this study consisted of patients referred to a neurology clinic at a tertiary center. Even those who had CNVs overlapping with well-known pathogenic CNVs or syndromes did not always show typical features [222324252627282930]. This could result partly from the different breakpoints among the patients, a combination of other genomic imbalances, or other individual genetic factors. As it is difficult to infer the pathogenic genome regions from phenotypes, genome-wide CMA is preferable to FISH for targeted detection of genomic imbalances [31]. Therefore, CMA testing should be conducted for every patient with DD/ID, regardless of other involved features [182021].
Among the cases with parental testing data available, most (86.4%) carried de novo CNVs. Why the proportion of de novo CNVs is rather high compared with other studies remains unclear [2132]. We could not investigate an association between de novo pathogenic CNVs and paternal age because data on the latter were not available [33]. Another potential reason is that we used stricter criteria for pathogenic CNVs and confirmed pathogenicity by parental testing. In 7.3% (8/110), the pathogenic CNVs were inherited from a healthy parent carrying an apparently balanced translocation. No familial aberrations were detected. We provided genetic counseling to those families at risk of recurrence. A higher than expected prevalence of parental balanced translocation might be a cause of pathogenic CNVs in patients with DD/ID [34], although CMA and FISH were not performed for all parents in the present study. If one deletion and one duplication involving two chromosomes, indicating unbalanced translocations, are simultaneously identified in a CMA test, the possibility of a balanced translocation in a parent should be considered.
We verified five rarely reported pathogenic CNVs: deletions on 2p21p16.3, 3p21.31, 10p11.22, 14q24.2, and 21q22.13. These genomic imbalances have rarely been reported in public databases or the literature [3536373839]. It is of value to expand the profile of genomic imbalances by confirming pathogenic CNV regions—together with the phenotype—that do not completely overlap with known pathogenic regions. Our study provided further evidence of pathogenicity, such as de novo status, for these five rarely reported CNVs, of which the pathogenicity remained uncertain without information of inheritance.
In conclusion, we report a relatively high diagnostic yield by CGH microarray analyses in a very large cohort of children with DD/ID, with results being confirmed by FISH/qPCR and parental testing in more than half of the cases, compared with other studies in Korea [7910]. We demonstrate the clinical diagnostic utility of CMA testing for pediatric populations in some countries, including Korea, where CMA testing has not been implemented in routine clinical settings for patients with DD/ID [2378]. CMA testing with appropriate resolution should be conducted for diagnostic use. Parental tests with validation studies should be conducted if possible because their results are critical for determining the clinical importance of novel CNVs and for providing genetic counseling.
 XML Download
XML Download