

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Mitochondria are generally characterized as the powerhouse of the cell, since this is the site where energy is produced from ATP. In addition to energy production, mitochondria play a key role in several important cellular processes, including growth, signaling, differentiation, reactive oxygen species (ROS) production, apoptosis, and cell cycle control. Interestingly, unlike other cellular organelles, mitochondria have their own DNA, mitochondrial DNA (mtDNA), and several studies have indicated an association between the accumulation of mtDNA mutations and mammalian aging [123].
Historically, mitochondria have not been considered important in restoring the functions of aged hematopoietic stem cells (HSCs); however, emerging studies on rejuvenating HSCs suggest an association between sirtuins (SIRTs) and mitochondrial activities [45]. In addition, a study on the deregulation of the mitochondrial stress-mediated metabolic system demonstrated that SIRT7 strongly influences the regenerative capacity of HSCs [6]. Although the functions of musculoskeletal stem cells (MuSCs) and HSCs are distinct, alteration of the SIRT1-associated nuclear/mitochondrial axis appears to be a common hallmark of aging in both cell types [78].
Recent research suggests the possibility of restoring the mitochondrial functions of aged stem cells, including MuSCs, nerve tissue stem cells (NSCs), and melanocyte stem cells (McSCs), by NAD+ supplementation without genetic manipulation [89]. The remedial effect of the NAD+ precursor nicotinamide riboside (NR) enhances mitochondrial functions in stem cells, including respiration, membrane potential, ATP production, and the mitochondrial unfolded protein response (UPR); however, these effects are not observed in stem cells with a SIRT1 deficit. Moreover, NR was found to suppress the process of senescence in adult NSCs and McSCs [8].
These findings have reinforced the notion that NAD+ precursors can function as a pharmacological tool to enhance SIRT activities. This, in turn, paves the way for clinical translation of NAD+ precursor treatment through further investigations of hematopoietic tissues. We review evidence relating mitochondrial dysfunction to HSC aging, and propose a strategy for mitochondrial-targeted recovery as a potentially safe, effective, and non-invasive method for the control or prevention of aging-related hematopoietic diseases.
ROLE OF THE MITOCHONDRIA IN HUMAN HEMATOPOIESIS
Mitochondria are central to the heme biosynthetic pathway, part of which occurs in the cytoplasm, eventually returning to the mitochondrion. Enzyme defects in the heme biosynthetic pathway cause sideroblastic anemia, leading to a deficiency of heme precursors and mitochondria that cannot fully utilize iron. In erythroid precursors, most of the iron initially gains access to the cell through transferring receptors, subsequently entering the mitochondria where it combines with protoporphyrin IX to produce heme. The heme produced leaves the mitochondria to merge with globin chains and synthesize cytoplasmic ribosomes [10]. Thus, when protoporphyrin synthesis is highly impaired, the imported iron instead accumulates in the mitochondria due to reactant deficiency (Fig. 1).
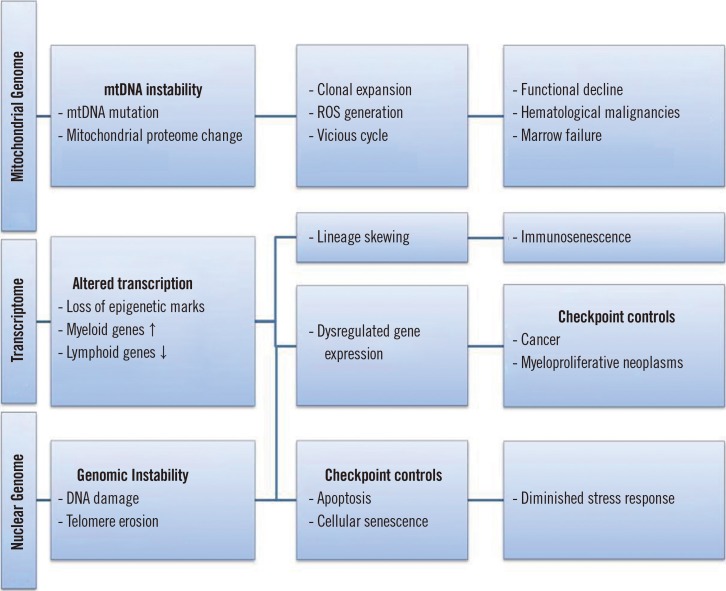
CONTRIBUTION OF MITOCHONDRIAL DYSFUNCTION AND GENOME ALTERATIONS TO HSC AGING
HSCs are generally dormant but have the potential to become highly active to restore blood on demand. Sustenance of HSC dormancy requires supply of low metabolic activity by glycolytic metabolites [1112]. Thus, unlike MuSCs, NSCs, and McSCs, which highly depend on mitochondrial ATP generation, the role of mitochondria in HSC homeostasis has traditionally not been emphasized.
During ATP production through oxidative phosphorylation, ROS are produced as the by-product of mitochondrial respiration [13]. Owing to their low metabolic activity, dormant HSCs exhibit very low levels of ROS that are closely associated with cellular metabolic activity [1214]. Accordingly, this unbalanced accumulation of ROS mediates HSC dysfunction [151617], and recent evidence implies that mitochondria play crucial roles in the maintenance of HSC quiescence and their capacity to switch from dormancy to a metabolically active state [18192021].
From a clinical perspective, HSCs are characterized by a very high turnover rate; however, they are not exempt from age-related insults. Aged HSCs are associated with increased incidence of myeloid proliferative diseases, such as marrow failure and hematopoietic neoplasms, and deterioration of the adaptive human immune system [22]. Because HSCs play a crucial role in maintaining the circulation, functional deterioration of HSCs may be highly responsible for age-related damage. Although HSCs maintain hematopoiesis for multiple processes, they are subject to drastic phenotypic and functional changes during aging, as confirmed by serial transplantation studies in mice [22]. The most notable of these changes is failure of the adaptive immune system, resulting in the weakened lymphoid function that is common in the elderly. In addition, aging often leads to the overproduction of myeloid cells, which fosters a pro-inflammatory hematopoietic environment [2223].
Aging leads to several clinical conditions related to the hematopoietic system, including decreased functionality of the adaptive immune system, elevated incidences of certain autoimmune diseases, age-associated anemia, and hematological malignancies [22]. Similar to other human tissues, the aged hematopoietic system experiences a decline in regenerative capacity for normal homeostasis after stress or damage [22]. Several pathways are involved in the aging mechanisms of the hematopoietic system, and both intrinsic and extrinsic factors are related to the aging environment (Fig. 2). However, recent studies suggest that age-dependent cellular and mitochondrial damage within the most primitive HSCs may play a crucial role in hematopoietic deterioration during aging [2425].
REJUVENATING AGED HSCS THROUGH RESTORING MITOCHONDRIAL DYSFUNCTION
One possible explanation for mammalian mitochondrial dysfunction during aging is the accumulation of altered nuclear and mitochondrial genetic materials, although environmental factors are clearly at play as well.
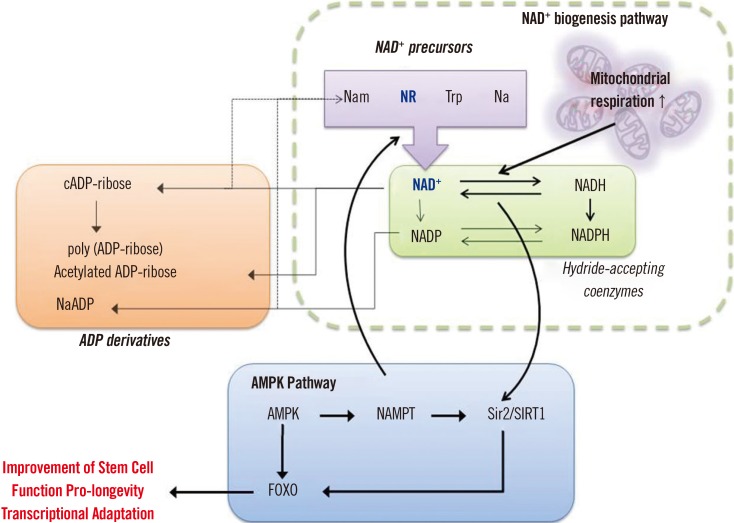
NAD was discovered over a century ago, and its role as a redox agent in metabolism has subsequently been established [26]. More recently, the oxidized form, NAD+, was revealed to be a key factor in mitochondrial function, and NAD+ supplementation has been shown to restore the normal phenotype [26]. Recent studies have clearly shown the role of NAD+ dietary supplements, commonly referred to as niacin or vitamin B3, in mitochondrial function, including tryptophan (Trp), nicotinic acid (Na), nicotinamide (Nam), and the newly identified NAD+ precursor NR [27282930].
More importantly, aging is related to SIRT deficiency and decreased mitochondrial function, and the NAD+/SIRT pathway is a pivotal factor in sustaining health and forestalling age-related diseases [31]. Indeed, SIRTs influence a broad range of cellular activities such as aging, transcription, apoptosis, and inflammation [32]. One study, using engineered mice expressing excess SIRT1, demonstrated that the level of cellular NAD+ gradually drops during normal aging [33]. Furthermore, NR was shown to enhance oxidative metabolism by increasing the NAD+ level and activating SIRT1 and SIRT3, suggesting the potential of NR as a pharmacological supplement to recover the metabolic and age-related disorders characterized by mitochondrial dysfunction [34].
Many studies have provided evidence indicating that SIRTs of the nucleus/mitochondrial control axis have age-specific effects in mediating the phenotypes of HSCs. Differentiation of SIRT1-deleted HSCs exhibited the typical characteristics of aged HSCs, such as a decline in the lymphoid compartment, anemia, and altered expression of related genes, indicating an essential role of SIRT1 in HSC homeostasis [7]. SIRT3 regulates the global acetylation of mitochondrial proteins and a stress response. Brown et al [4] showed that SIRT3 expression was suppressed with aging, and upregulation of SIRT3 enhanced the regenerative capacity of aged HSCs. When SIRT7, which controls the de-acetylation promoter, was inactivated in HSCs, quiescence was reduced and the mitochondrial protein folding stress (PFSmt) occurred, ultimately resulting in compromised regenerative capacity. By contrast, SIRT7 up-regulation enhanced the regenerative capacity of aged HSCs [6]. Another recent study also showed that SIRT6-deficient HSCs exhibited impaired self-renewal capacity [27].
POSSIBLE APPROACH FOR CORRECTING AGING-RELATED HSC DYSFUNCTION
Based on the studies described above, we propose a mitochondria-targeted strategy for controlling the HSC aging mechanism and associated regulatory factors toward restoration of aged HSC function through improving mitochondrial function (Fig. 3).
The aging process of HSCs is potentially linked with poly ADP-ribose polymerase (PARP) activation, NAD+ deficiency, SIRT inactivation, mitochondrial dysfunction, and cell and tissue damage, reflecting a generalized aging syndrome, which may be corrected by supplementation with NAD+ precursors (Fig. 4). In particular, recent studies have indicated that NR supplementation could rejuvenate aged stem cells such as MuSCs, NSCs, and McSCs, enhance lifespan, and improve muscle function in muscular dystrophy models [89]. Moreover, the association of SIRT1 with the aging-like phenotypes of both MuSCs and HSCs suggests a plausible pharmacological approach by targeting the interplay of SIRTs and the nuclear/mitochondrial control axis in HSCs [4578].
An important consideration is to examine the feasibility of activating SIRT3, SIRT6, and SIRT7, which have specific rejuvenation effects on HSCs [456]. For example, experiments with SIRT1-deficient mice provided NR supplementation in tablet form showed improvement of muscular dystrophy through enhancing mitochondrial function [89]. This implies that NR would easily access human hematopoietic tissues to exert the expected rejuvenation effect of aged HSCs.
Moreover, NR supplementation in mammalian cells increases NAD+ levels and activates SIRT1 and SIRT3, leading to amelioration of the metabolic and age-related degeneration characterized by mitochondrial dysfunction [34]. Therefore, more detailed studies on the effects of NR, the NAD+ precursor, on SIRTs to improve the mitochondrial activities of HSCs may lead to the development of optimal strategies for reversing age-dependent degeneration with an accessible approach.
CONCLUSION
Although further research on the effects of NAD+ replenishment in human health maintenance is required, the studies we reviewed clearly demonstrate that the level of NAD+ drops during the aging process [37]. In addition, the relevant pathways of NAD+ synthesis, SIRTs, and PARPs suggest that NAD+ replenishment may be beneficial in rejuvenating aged HSCs.
The ability to rejuvenate stem cells without relying on genetic manipulation is the safest way to achieve optimal clinical outcomes. Regulation of aging through enhancing mitochondrial function is a potentially effective, low-cost, and stable treatment method.



 XML Download
XML Download