

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Ovarian cancer is the leading cause of mortality among all gynecological malignancies, with 238,700 new cases and 151,900 deaths worldwide [12]. In China, 52,100 new cases and 22,500 deaths were predicted in 2015 [3]. Although the treatment for ovarian cancer is constantly evolving, the overall survival (OS) rates have not improved significantly.
Recent advances in our knowledge of the molecular traits underlying ovarian cancer not only contribute to the identification of familial cancer predisposition but also address potential predictive biomarkers and therapeutic alternatives such as anti-angiogenic agents (Bevacizumab) and poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) [4]. The PARP-1 protein is critical for the repair of single-strand DNA breaks. In cells with defective homologous recombination (HR) such as the BRCA1/2-mutation carriers, PARP-1 inhibition is lethal and results in cell cycle arrest and subsequent apoptosis [5].
Inherited BRCA1/2 mutations account for the majority of familial ovarian cancer [6] and are among the most frequently mutated genes in high-grade ovarian serous carcinomas, which are responsible for the vast majority of ovarian cancer deaths [78]. BRCA1/2-associated ovarian carcinomas show improved OS and sensitivity to both platinum chemotherapy and PARPi [910111213141516]. In 2014, Olaparib (PARPi) was granted approval by the Food and Drug Administration (FDA) for the treatment of patients with BRCA1/2-mutated ovarian cancer who have been treated with 3 or more prior lines of chemotherapy, and the European Medicines Agency (EMA) approved Olaparib for maintenance treatment of patients with BRCA1/2-mutated platinum-sensitive relapsed high-grade serous ovarian carcinomas who are responsive to platinum-based chemotherapy.
PARP inhibitors are active in patients with germline BRCA1/2 mutations as well as in a subset of “sporadic” recurrent platinum-sensitive ovarian carcinomas [12]. This may be attributed to ATM, BARD1, NBN, and other genes in the Fanconi anemia (FA) pathway, which play key roles in HR, the primary mechanism that repairs double-strand DNA breaks (DSBs) [7]. In vitro studies demonstrate that defects in other HR proteins such as ATM, CHEK1, CHEK2, NBN, and RAD51D also confer sensitivity to PARPi [1517]. Somatic BRCA1/2 mutations and alterations in other HR genes have a similar positive impact on OS and platinum responsiveness as those of germline BRCA1/2 mutations [18]. Specifically, the Cancer Genome Atlas (TCGA) reported HR deficiency in approximately 50% of high-grade serous ovarian carcinomas [7]. Moreover, germline or somatic mutations in HR genes are present in both serous and non-serous histologies [18]. Though germline detection of BRCA1/2 is currently in extensive genetics testing, this approach does not allow patients with somatic and other HR gene mutations to avail themselves of the opportunity to use DNA-damaging agents.
Herein, we applied high throughput next-generation sequencing (NGS) technologies to test DNA from 50 paired whole blood samples and frozen tumors. Both germline and somatic deleterious mutations, covering 31 core HR genes, were identified in Chinese women with epithelial ovarian carcinomas (EOC), and the relationship between genetic alterations and clinical parameters was analyzed.
MATERIALS AND METHODS
1. Patients and samples
A cohort of 50 women with EOC who underwent surgical resection between 2010 and 2015 (median age, 53 years) at the Peking Union Medical College Hospital (PUMCH) were consecutively included in the study. Patients were prospectively enrolled at diagnosis and not selected for age, familial cancer history or histological subtype. Clinical information was retrieved from medical records: 48 patients received chemotherapy at our institution and were routinely followed in our outpatient department. The diagnosis for the case of simultaneous ovarian and endometrial cancer was confirmed under consultation with dedicated gynecological oncologists and pathologists after review of biopsy sections. Informed consent was provided by patients and the study was approved by the Institutional Review Board (IRB) of PUMCH, Beijing, China. The data of all patients were analyzed anonymously, and for research use only.
Genetic tests were performed after pathological diagnosis of EOC. Genomic DNA (gDNA) was extracted from peripheral blood mononuclear cells (germline) and from frozen sections in areas with a minimum neoplastic cellularity of 70% (somatic). DNA quantification, library construction, hybridization and NGS of the whole exome were performed by the BGI group (Shenzhen, China). Briefly, gDNA was extracted and quantified using Qubit (Life Technologies, Gaithersburg, MD, USA). The gDNA was fragmented randomly by Covaris, and after 2 rounds of bead purification, the resulting gDNA fragments should be distributed mainly between 200 and 400 bp. The AdA adaptor-ligated fragments were amplified by polymerase chain reaction (PCR) and the products were used for follow-up exon capture (BGI Exome V4 Kit, 59M; BGI group). Captured fragments were subsequently purified, amplified, ligated with AdB and circularized. Finally, high-throughput sequencing of library products was performed by Complete Genomics (CG) Black Bird platform (BGI group). All the exons of each target gene were sequenced.
2. Variant calling and bioinformatics analysis
Data analysis, including base calling, alignment to the reference human genome (UCSC hg19) and variant calling, was done using domestically developed suite software based on the CG platform (BGI group). Filtered single-base substitutions, small insertions and deletions (indels) were annotated using SnpSift and the National Center for Biotechnology Information (NCBI) dbSNP database (v141). Loss-of function mutations were validated by Sanger sequencing.
Selected genes included BRCA1/2, core genes in the FA pathway (C19orf40, FANCA, FANCB, FANC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, PALB2), HR RAD genes (RAD50, RAD51, RAD51C, RAD51L1, RAD51L3, RAD52, RAD54B, RAD54L) and other DNA damage response genes involved in HR (ATM, ATR, CHEK1, CHEK2, BARD1, BRIP1, FAM175A, MRE11A, NBN) [4718].
Each variant was annotated with respect to gene location and predicted function in Human Genome Variation Society (HGVS) nomenclature. Variants in BRCA1/2 were defined as deleterious if they were recorded as pathogenic or likely pathogenic in ClinVar (for germline mutations) and COSMIC (for somatic mutations). Missense, nonsense mutations and frameshift indels in BRCA1/2, though not recorded or annotated with uncertain/conflicting significance in databases, were counted as pathogenic if they resulted in protein truncation in functional domains. For HR genes other than BRCA1/2, nonsense, disruptive inframe and frameshift indels were defined as loss-of-function whereas only missense variants previously demonstrated to be deleterious were included.
3. Statistical analysis
Categorical variables were described with a frequency value, and continuous variables with the median and range. Risk factors were explored univariately between groups using log-rank tests or Fisher's exact tests, with relative risk (RR), and 95% confidence intervals (CI) calculated. Multivariate analysis was conducted using the logistic regression method. The 1st degree family history referred to hereditary breast and ovarian cancer (HBOC) and the 2nd degree family history included lung, esophagus, stomach, pancreas, intestine, and endometrial carcinomas in the patients themselves or in their immediate relatives [19]. Platinum sensitivity was defined as the maintenance of complete remission of 6 months or more after the completion of frontline chemotherapy. The observation time for OS ranged from the date of diagnosis to the date of death or the study end date, whichever occurred first. The endpoint for progression-free survival (PFS) was either the date of first recurrence or the last follow-up, starting from the completion of frontline chemotherapy. Potential impact factors for prognosis were analyzed univariately using the Kaplan-Meier method. Multivariate analysis for survival was done using a COX regression model with hazards ratio calculated. A p-value <0.05 (2-sided) was considered statistically significant. Statistical analysis was performed using Statistical Product and Service Solutions (SPSS) Statistics 20.0 (IBM Corporation, Armonk, NY, USA).
RESULTS
Fifty Chinese women diagnosed with EOC were included in this study. Table 1 and Supplementary Table 1 depict the major clinical characteristics of these patients. The majority of tumors (39/50, 78%) were high-grade serous carcinomas (HGSC), though histological subtype did not limit enrollment. Five patients had 1st degree family history: 2 patients previously had breast cancer themselves, and more than one first-degree relative of the other 3 patients had HBOC. In addition, 6 cases had 2nd degree family history. Eighty percent of the cases were in the advanced phase when diagnosed and nearly 80% were optimally cytoreduced by surgery (maximal residual tumor diameter <1 cm).
Table 1
Clinical characteristics and pathogenic HR mutations

DNA from all samples was successfully amplified in multiplex PCR and an adequate library for NGS was obtained. The average depth on targets was 169.2, with 95.3% target bases covered more than 10 times. We identified 169,151 single-nucleotide variants (SNVs) and 18,544 insertions/deletions using whole exome sequencing based on NGS.
1. Overall deleterious HR mutations
Eighteen subjects (36%) had 21 deleterious germline mutations in 7 different HR genes (i.e., ATR, BRCA1/2, CHEK2, RAD50, RAD52, and RAD54B), all with corresponding somatic mutations. There were 2 recurrent deleterious germline mutations: RAD52, c.1037C>A and RAD54B, c.1778A>G (Fig. 1B). Another 5 subjects (10%) harbored only somatic mutations, one for each patient, in BRCA1/2, RAD50, and RAD54B (Fig. 1C). These somatic mutations were tumor-specific, and were not identified in the corresponding blood samples. There was no retroversion in the somatic and germline analysis. Three cases each possessed 2 germline mutations in different HR genes (Table 2). Therefore, the total proportion of subjects with at least one pathogenic germline or somatic HR mutation was 46% (Fig. 1A). Of all 24 deleterious mutations, 5 (20.8%) occurred in BRCA1, 9 (37.5%) were in BRCA2, and 10 (41.7%) were in other HR genes: ATR, CHEK2, RAD50, RAD52, and RAD54B. Table 2 lists the loss-of-function mutations identified in HR core genes detected using NGS from the 50-matched whole blood and frozen tumor samples. Moreover, none of the clinical features listed in Table 1 and Supplementary Table 1 have been proven to be associated with HR mutational status (p>0.05).
Fig. 1
(A) Overall pathogenic mutations in HR genes. (B) Gene distribution of germline HR mutations. (C) Gene distribution of somatic HR mutations. Values are presented as number (%).
HR, homologous recombination.
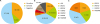
Table 2
Pathogenic mutations in HR core genes detected using NGS of 50 paired ovarian cancer samples

2. BRCA1/2 and other HR deleterious mutations
Of the 5 mutations in BRCA1, 3 are frameshifts and 2 are nonsense SNVs. One frameshift alteration (c.3359_3363delTTAAT) is recorded as a “variant of uncertain significance” in ClinVar but results in protein-coding truncation before the BRCA1 C Terminus (BRCT) domain, which is predominantly involved in cell cycle checkpoint functions responsive to DNA damage [20]. Therefore, this mutation is considered deleterious in the following analyses. The remaining frameshift deletions and nonsense mutations (4/50, 8%) are recorded as pathogenic or likely pathogenic in ClinVar or COSMIC database.
Of the nine BRCA2 mutations, 5 are frameshifts (3 deletions and 2 insertions) and 4 are SNVs. Two of the frameshifts and the only somatic point-mutation (nonsense) in BRCA2 have not been recorded yet, but are considered pathogenic as they cause a premature stop codon, a feature of pathogenicity. The remaining one nonsense, 2 missense mutations and 3 frameshift indels (6/50, 12%) are recorded with pathogenicity in ClinVar (germline).
Unlike BRCA1/2, the clinical significance of other HR-gene variants has not been well annotated in databases. In our analysis, only one germline point missense mutation (RAD54B and c.1778A>G) was previously demonstrated to be pathogenic (ClinVar). However, nonsense mutations (2), disruptive inframe insertions (2), and frameshifts (5) in other HR genes are considered deleterious.
Collectively, BRCA2 was mutated most frequently among the selected 31 HR core genes, whereas the majority of studies found that BRCA1 was the predominant pathogenic gene [791016]. BRCA1/2 mutations occupied 57.1% of the germline alterations whereas 3 out of the 5 somatic mutations were in RAD genes (Fig. 1).
Deleterious BRCA1/2 mutations were closely related to family history, specifically the HBOC syndrome (p<0.05). However, when other HR genes and 2nd degree family history were taken into account, these relationships weakened (p>0.05, Table 1). Only one case (9.1%) from the non-serous tumors had a BRCA1 mutation whereas 13 of 39 (33.3%) serous cases had a deleterious germline or somatic BRCA1/2 mutation, though the difference was not yet statistically significant. A wider distribution of non-BRCA1/2 HR genes was discovered altered in non-serous histologies: in the non-serous cases, 4 of the 5 mutations were in genes other than BRCA1/2 (one subject had 2 pathogenic mutations). In contrast, only 38.1% (8 of 21) of pathogenic mutations in serous carcinomas were in other HR genes. Moreover, loss-of-function HR mutations were identified in 2 of 3 endometrioid (sample T11, T32) and 2 of 2 mucous carcinomas (sample B4, B34). No HR defects were found in clear cell carcinomas (CCC) and transitional cell carcinomas (TCC).
3. Remission rate and primary platinum response
Forty-eight (96%) patients had adequate available clinical information to define primary platinum response, with 2 cases receiving their chemotherapy back at local hospitals. Thirty-six cases were platinum-sensitive defined as the maintenance of complete remission of 6 months or more after the completion of frontline chemotherapy. Forty-three out of 48 cases (89.6%) achieved complete remission after initial cytoreductive surgery and normative platinum-based chemotherapy. The presence of a germline or somatic mutation in BRCA1/2 was closely associated with primary platinum sensitivity, with an RR of 1.57 (95% CI=1.22–2.00; Fig. 2). All 13 cases (100%) with an identified BRCA1/2 mutation demonstrated platinum sensitivity. In contrast, 23 of 35 (65.7%) patients without a BRCA1/2 mutation harbored platinum responsiveness and the remainder were either refractory or platinum-resistant. Considering HR mutations that were not restricted in BRCA1/2, the predictive ability of genetic alterations for chemo-response was lost. Unexpectedly, all 3 carcinomas with somatic mutations in other HR genes showed platinum resistance whereas the germline mutations indicated similar sensitivity to platinum than the group without any HR variant. Univariate analysis revealed that platinum sensitivity was also impacted by optimal cytoreduction (p<0.05).

4. OS and relapse
With a median follow-up duration of 24.6 m (9.1–69.5 m) from initial diagnosis, 5 patients died due to EOC. The estimated 5-year OS was 83.6% using Kaplan-Meier method, with a median survival period of 61.7 m. The presence of a pathogenic mutation in non-BRCA1/2 HR genes was associated with poorer OS (p<0.05, Fig. 3A): patients with mutations in other HR genes had a median survival of 30.6 m, compared with 63.4 m for patients without any HR variant. Moreover, the cases (100.0%) with germline or somatic BRCA1/2 mutations were all alive, who had similar OS as cases without deleterious HR mutations (p>0.05). The stage, platinum sensitivity and initial remission status were also found significantly related to OS (p<0.05) using univariate analysis.
Fig. 3
Overall survival impacted by (A) HR mutations and (B) platinum sensitivity.
HR, homologous recombination.

In a multivariate model including the above 4 impact factors, only the chemo-responsiveness remained significantly associated with OS (p<0.05). Median OS in patients with resistance to platinum was 41.8 months, which was significantly worse than the 66.9 months for cases with sensitivity to frontline chemotherapy (HR=0.053; 95% CI=0.004–0.774) (Fig. 3B).
Although the proportion of platinum-sensitive cases reached 75% in the study cohort, leading to a remission rate greater than 80%, 62.8% of initially remitted cases had recurrence. The median PFS interval was merely 19.3 months, predicted by the Kaplan-Meier method. Subjects without any deleterious mutation in HR genes had worse PFS (15.7 m) than BRCA1/2-mutation carriers (25.9 m) and patients with other HR mutations (19.2 m, p>0.05, Fig. 4A). However, patients with a somatic mutation in other HR gene had an especially poor PFS of 5.5 m. Instead of genetic alterations, PFS was significantly influenced by surgical thoroughness and chemo-sensitivity (p<0.05, Fig. 4). Multivariate analysis revealed that suboptimal cytoreduction was the only independent predictor for relapse (HR=0.247; 95% CI=0.083–0.739; Fig. 4B).

DISCUSSION
As expected, loss-of-function BRCA1/2 mutations were strongly associated with HBOC, confirming the fact that risks for breast and ovarian cancer increase in BRCA1/2-mutation carriers [21]. Therefore, deleterious BRCA1/2-mutation carriers and their close family members could benefit from genetic counseling and prophylactic risk-reduction treatment under certain indications [22]. On the other hand, of women with inherited mutations, 9%–30% had no reported family history of breast or ovarian cancer [62324]. The proportion might be underestimated due to limited genetic testing in patients without a strong family history. In addition, BRCA1/2 mutation carriers have an increased RR for prostate, pancreas, endometrium and cervical cancer, ductal gall, stomach, and skin cancer [25262728]. In our cases with a 2nd degree family history, deleterious BRCA1/2 mutations were discovered in one third of them. Therefore, germline BRCA1/2 testing is currently recommended for all women diagnosed with EOC regardless of family history by the Society of Gynecologic Oncology (SGO), the National Comprehensive Cancer Network (NCCN), and other academic societies.
Despite familial predisposition, BRCA1/2-associated ovarian carcinomas reveal higher response rates to platinum-based treatment, longer interval between relapses, and improved OS [2930]. All our BRCA1/2-mutation carriers were platinum-responsive and alive during surveillance. In addition, patients with platinum-sensitive recurrent serous ovarian cancer with a BRC1/2-mutation have the greatest likelihood of benefiting from PARPi (e.g., Olaparib), by inducing synthetic lethality in tumors with HR deficiency [3132]. There is emerging evidence of abnormalities in HR genes other than BRCA1/2, including germline and somatic mutations in ATM, BARD1, BRIP1, CHEK1, CHEK2, FAM175A, MRE11A, NBN, PALB2, RAD51C, and RAD51D [46718]. We found that approximately 20% of our patients harbored at least one non-BRCA1/2 HR mutation, and their clinical features differed from BRCA1/2-mutation carriers to some extent. The TCGA Research Network has reported that up to 50% of patients with high-grade serous ovarian cancer are deficient in HR due to germline or somatically acquired BRCA1/2 mutations, epigenetic inactivation of BRCA1, or BRCA1/2-independent defects in the HR pathway. In addition, loss-of-function mutations in HR/FA core genes predict a higher rate of platinum sensitivity and better OS, which is consistent with in vitro data that cells with defective HR are more sensitive to agents that induce DSBs [518]. The copy number deletion of RAD50 was revealed as a candidate marker for survival and response to PARPi in BRCA1/2 wild ovarian tumors [33]. Interestingly, we also identified RAD genes were the most frequently mutated in all somatic mutations. These findings support the hypothesis that HR deficiency might be the Achilles heel of this deadly disease, yet the most established BRCA1/2-associated mechanism only accounts for approximately 15% of OC patients [34]. Therefore, further investigation to complete the molecular image of the HR pathway in tumor genesis and development would help: 1) better understand tumor heterogeneity, which have been largely ignored by standard treatment strategies; and 2) judge if PARPi also has a therapeutic effect on the remaining BRCA1/2 wild-type patients.
Though BRCA1/2 are the dominant mutated genes in HGSC, we found HR gene (germline and somatic) alter not rarely in non-serous histological subtypes. Mutations in HR genes were present in 36.4% non-serous carcinomas, including mucous and endometrioid tumors. Similarly, though non-serous cases had some BRCA1/2 mutations, they had a greater proportion of mutations in other HR genes [18]. Non-serous ovarian carcinomas might also have a meaningful risk of hereditary cancer but identification necessitates assessment with a larger panel of ovarian cancer susceptibility genes. Conversely, certain guidelines (e.g. Scottish Intercollegiate Guideline Network) recommend genetic testing to be offered merely for non-mucinous EOC [35]. In addition, some researchers identified pathogenic BRCA1/2 mutations in 8.4% of women (10 of 119) with endometrioid ovarian carcinoma and in 6.3% (4 of 63) of women initially reported to have CCC or mixed CCC/serous ovarian carcinoma. However, the pathological diagnosis for some of these cases was revised after immune-histopathology review and genomic analyses and they concluded that BRCA1/2 germline mutations are almost exclusively associated with HGSC [9]. Therefore, we suggest more deliberate clinical trial design as most recent trials focused on merely HGSCs. Molecular pathological diagnosis might be more precise for optimizing treatment schemes for individual patients with tumor heterogeneity.
We acknowledge that there are several limitations in the present study. With a relatively small sample size, any conclusion should be declared with caution. Mutation carriers in HR genes other than BRCA1/2 manifested different but inconclusive clinical characteristics. For instance, non-BRCA1/2 HR mutations appeared to have an inverse effect on prognosis whereas findings from other research suggested that HR mutations indicate better survival. In addition, NGS presents high throughput and sensitivity in detecting alterations but generates a large amount of complicated data that require bioinformatics analysis [36]. Specifically, annotations of variants in HR genes other than BRCA1/2 are seriously inadequate, requiring further functional experiments and pedigree analysis for validation [37]. Integral databases, such as the Breast Cancer Information Core (BIC), are also needed for interpretation of these results.
In conclusion, 36% and 10% of the unselected cases with EOC possessed deleterious germline and tumor-specific HR-gene mutations, regardless of age, family history, and histology. BCRA1/2, other HR-gene, and none mutation carriers presented different clinical features. Panel genetic testing of germline and somatic HR mutations is recommended to identify a comprehensive profile of HR defect and to elucidate its potential value in target therapy.
 XML Download
XML Download