

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Guillain-Barré syndrome (GBS) is the most common cause of acute polyneuropathy. The clinical-pathologic spectrum of GBS extends from the classical acute inflammatory demyelinating polyneuropathy (AIDP) to pure axonal variants with (acute motor sensory axonal neuropathy) and without (acute motor axonal neuropathy) sensory involvement, and clinical variants such as the Miller Fisher syndrome (MFS). Cranial-nerve involvement, dysautonomia, and respiratory insufficiency may be seen during the course of the disease.1,2 Although the cranial nerves are often involved in GBS, the optic nerves are usually spared, presumably because they are part of the central nervous system (CNS).3
A few studies have revealed optic-nerve involvement and evoked potential abnormalities in GBS.4-7 In the present study we determined the incidence of visual pathway involvement in GBS with the aid of clinical and electrophysiological assessment, and defined the patterns of visual evoked potential (VEP) abnormalities in GBS.
Methods
Thirty-two patients with a diagnosis of GBS at the Department of Neurology, Ondokuz Mayıs University Health and Research Hospital between April 2005 and December 2009 were included in the study. All patients met the diagnostic criteria for GBS, as defined previously, based on clinical evaluation, nerve conduction studies, and cerebrospinal fluid (CSF) investigation.8-10 All patients had symptomatic motor or sensory neuropathy with acute onset. Electrophysiological data were consistent with demyelinating, axonal, or mixed polyneuropathy; no other etiology of acute neuropathy was detectable. All patients gave their informed consent to participate in the study. Patients with severe motor, bulbar, or autonomic involvement causing cardiopulmonary instability and needing intensive life support and monitoring, or who died early during the course of illness were excluded.
The neurologic findings of each patient were outlined in six categories with the aid of neurologic examination:
Superficial (any deficiency of light-touch, pinprick, or temperature sensation) and deep (vibratory sensation or position sense) sensorial loss.
Motor deficit (decrease in upper or lower extremity muscle strength).
Presence of limb ataxia.
Cranial-nerve involvement (other than the optic nerve).
Autonomic involvement.
Routine blood tests and CSF examination were performed. The clinical syndrome was defined according to electrophysiological and clinical findings.
A pattern reversal-VEP study was carried out for all patients as early as possible when clinical cooperation with the test was technically available. The stimulation source was a black/white full-field checkerboard pattern on a television screen with check size of 14 inches, a reversal rate of 1 Hz, and a Michelson contrast of 99%. The television screen was positioned 1 m from the eyes. Each eye was tested separately and with the opposite eye occluded. The VEPs were recorded by epicranial surface electrodes. The active electrode was placed over the midocciput (Oz) and referred to as the midfrontal lead (Fz). The ground electrode was placed at the vertex (Cz). A bandpass filter (0.1-1 Hz) was used with a sweep speed of 300 msec.11,12 In total, 375 responses were recorded for each eye and averaged by a computer system (Dantec Keypoint, Medtronic Functional Diagnostics, Skovlunde, Denmark). Two trials were performed under the same stimulation conditions for each subject to confirm the reproducibility. The latencies and amplitudes of the N75, P100, and N145 waves, the P100 morphology, and differences in the latency and amplitude of the P100 wave between the two eyes were evaluated. The latencies and amplitudes were evaluated according to normal values from our laboratory obtained from 160 healthy subjects (114 females and 46 males) aged between 19 and 72 years. The following were considered to be abnormal: P100 latencies >2.5 standard deviations above the mean of the normal population (>108 msec for patients younger than 50 years, and >116 msec for males and >109 msec for females older than 50 years), minimal left-to-right amplitude ratio >0.66, and left-right difference of latency >6 msec for the P100 peak.
Each patient underwent a detailed ophthalmologic examination performed by an ophthalmologist who was blinded to the VEP results. Anterior segment evaluation, visual acuity, presence of pupillary light-reflex abnormalities (total loss, anisocoria, or relative afferent pupillary defect), fundoscopic findings, and defects of colored vision were recorded. Patients who had diseases that may affect VEP results, such as severe refractive error, glaucoma, optic media opacity, retinal disease, or previous history of optic neuropathy, were excluded.
Any correlations between the presence of pathologic VEPs and any category of neurologic deficit, electrophysiological data, or CSF protein level were examined. Fisher's exact, Student's t, and Mann-Whitney U tests were used for statistical analyses.
Results
The patients ranged in age from 19 to 77 years (mean±SD: 50.13±16.02 years). There were 19 males (59%) and 13 females (41%). The diagnosis was AIDP in 18 patients (56%), MFS in 5 (16%), acute motor sensory axonal neuropathy in 5 (16%), and acute motor axonal neuropathy in 4 (13%), based on clinical and electrophysiological findings. Twenty-one patients (66%) were treated with intravenous immunoglobulin and 4 (13%) were treated with plasmapheresis.
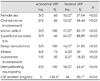
The time interval between the onset of symptoms and the VEP study ranged from 6 to 45 days. VEPs were abnormal in five cases (15.63%) (Fig. 1). The most common abnormality was increased interocular latency difference (7-20 msec), which was present in all five cases. P100 latency was delayed in four cases (12.50%), of which two had prolonged P100 latency in both eyes (6.25%). Other abnormalities were distorted (W-shaped) P100 configuration and increased interocular amplitude difference (two cases, 6.25%) (Table 1). N75 and N145 were significantly prolonged in only one patient (3.13%). Visual examination was abnormal in four of the five patients with abnormal VEPs (80%). Of those with abnormal VEPs, there was decrease in visual acuity in four patients (80%), light-reflex abnormalities or afferent pupillary defect in three (60%), dyschromatopsia in one (20%), and papilledema in one (20%) (Table 1). Among those with normal VEPs, only one patient had light-reflex abnormality (3.13%), and two had unexplained decreases in visual acuity (6.25%). One patient with prolonged P100 in one eye had unexplained decreased visual acuity and dyschromatopsia in the contralateral eye.
Three of the patients with abnormal VEPs were female (60%), four had a diagnosis of AIDP (80%), and one was diagnosed with MFS (20%) (Table 1). Pathologic VEPs were obtained only in patients with electrophysiological findings suggesting demyelinating peripheral neuropathy, but not axonal forms. None of the clinical deficits observed on neurologic examination was significantly correlated with VEP pathology (Table 2). Superficial sensorial deficit (60% versus 88.89%) and cranial-nerve involvement other than the optic nerve (40% versus 44. 44%) appeared to be less prevalent, and autonomic involvement (20% versus 18.52%) slightly more common in GBS patients with abnormal VEPs, but the difference did not reach statistical significance (p>0.05). All of the patients with pathologic VEPs had deep sensorial loss and motor involvement, while among those with normal VEPs, 51.85% had deep sensorial loss and 85.19% had motor loss (p>0.05). Limb ataxia, which could be evaluated in 24 patients, was more common in those with abnormal VEPs, but the difference was not statistically significant (75% versus 30%, p>0.05). CSF protein levels tended to be higher in GBS patients with pathologic VEPs, but again the difference did not reach statistical significance (133.2 versus 85.70 mg/dL) (Table 2).
Discussion
GBS is an acute, rapidly progressive, symmetrical polyradiculoneuropathy that is characterized by weakness, areflexia, sensorial loss, and albuminocytologic dissociation in the CSF. Ataxia and dysautonomia may also be seen.1,6 Cranial-nerve involvement may form part of the disease, especially in MFS. Ophthalmoparesis, facial weakness, or bulbar paralyses are common in patients with GBS, but optic-nerve involvement is less common.
Morley and Reynolds first drew attention to the probability of optic neuritis in GBS in 1966.13 Behan subsequently reported a case of GBS with bilateral decreased visual acuity to 20/70, dyschromatopsia, and optic disc swelling.14 Later clinical series and case reports have identified patients with GBS accompanied by optic neuritis. In the study of Mori, which included 45 patients, 42% had mydriasis and light-reflex abnormalities, and approximately 50% had anisocoria.15 Decreased visual acuity,7,16,17 total blindness,18 RAPD or other pupillary dysfunctions,17,19-22 dyschromatopsia,7,14,20,23 enlarged blind spot,7 centrocecal scotoma,16 temporal peripheral field constriction,21,24 and optic disc swelling16,17,25-27 have been described in patients with GBS. Some cases with optic neuritis may also have cerebral parenchymal abnormalities on MRI.28,29
Pathologies of the optic pathway are best demonstrated by VEP evaluation. The incidence of VEP abnormalities has been investigated in chronic inflammatory demyelinating neuropathy, and reportedly varies between 44% and 86%.30-32 Four previous studies examined the incidence of VEP abnormalities in GBS patients. Topçu et al.7 described VEP abnormalities in 33.33% of GBS patients in the pediatric age group. Zgorzalewicz et al.5 found elongation of P100 or N145 in 5 (17%) of 30 patients diagnosed with GBS between the ages of 8 and 18 years. In addition to these cases, Wong et al.6 found no VEP abnormality in his four pediatric cases, and only one with light-reflex abnormality. Finally, Durand et al. found a VEP abnormality in only one of nine adults with MFS (11%).4
In the present study we found abnormal VEPs in 16% of patients diagnosed with GBS. This rate is similar to that found for a pediatric group and adults with MFS. To our knowledge, the present study is the largest trial demonstrating VEP pathologies in adult GBS patients. Previously reported VEP abnormalities in GBS patients are absent VEPs,28 P100 elongation,7,20,24,27,33,34 N145 latency elongation,5 and alterations in P100 morphology.25 P100 latencies differed significantly between the two sides in all of our patients with pathologic VEPs, suggesting that if it is present, optic-nerve or postchiasmal involvement is always asymmetric. The other VEP abnormalities noted among our patients were elongation of the P100 latency in most of those with normal N75 latencies, distorted P100 configuration (W shaped), and pathologic differences in amplitudes between the two sides.
Nearly all of the patients in our study with abnormal VEPs had abnormal findings in ophthalmologic examinations: decreased visual acuity, RAPD, loss of light reflex, dyschromatopsia, and papilledema. One of our patients had light-reflex abnormality but normal VEPs. Fuller et al.35 also described a patient with severe demyelinating GBS and unreactive pupils, but with normal VEPs and a microscope examination revealing no demyelination in the optic nerve. It is thus impossible to conclude that all patients with pupillary dysfunction have prechiasmatic optic-nerve involvement. The N75, P100, and N145 waves recorded during our VEP study are known to originate from the striate cortex.36 Light-reflex abnormalities accompanied abnormal P100 latencies in 60% of our patients, suggesting involvement of the optic nerve. For a diagnosis of optic neuritis, light-reflex abnormalities must be accompanied with other ophthalmologic findings, such as decreases in visual acuity or dyschromatopsia and VEP abnormality. Our results also show that VEPs may be abnormal in GBS cases without visual complaints or ophthalmologic findings, suggesting the presence of central lesions in the visual pathways; however, the presence of brain lesions was not investigated in the present study.
Involvement of the second cranial nerve in GBS may be related to infectious agents such as Mycoplasma pneumoniae,17,18,25,34 cytomegalovirus,3 Epstein-Barr virus,28 mumps virus,37 and herpes simplex virus type I.20 Isolated optic neuritis may develop after mycoplasma infection.38 This correlation with infectious agents was not investigated in our study.
The cases reported in the literature give the impression that optic-nerve involvement accompanies MFS.7,19,23,24,26,29,39 The human optic nerve contains high levels of sulfated glucuronyl glycolipids and gangliosides such as GD1b, GQ1b, and GT1b.40,41 The involvement of both peripheral and optic nerves in GBS may result from these shared pathogenic epitopes.21,42 The amount of CNS involvement is greater in MFS than in GBS. Optic-nerve involvement may be part of a spectrum of CNS involvement.43 We were unable to find a significant association with optic pathway involvement and the MFS variant of GBS, although deep sensorial loss and limb ataxia were more prevalent among our patients with abnormal VEPs.
None of our patients with axonal GBS had abnormal VEPs, which may be attributable to the low sensitivity of the VEP study to axonal damage in the optic nerve. Although not statistically significant, the involvement of the motor and autonomic nervous systems was more prevalent in our GBS patients with abnormal VEPs, while superficial sensorial loss and involvement of other cranial nerves were less common. In addition, there was no significant correlation between CSF protein levels and VEP abnormalities. These differences between groups might not have reached statistical significance due to the very small number of patients with pathologic VEPs evaluated in the present study. It should also be emphasized that patients with severe deficits or who are early along the course of the illness were not included in this study.
VEPs may be abnormal in GBS, but this is not a frequent occurrence. The findings of our study underline the possibility of visual pathway involvement in GBS. If present, visual pathway involvement is always asymmetric. VEPs may be abnormal in only the demyelinating forms of GBS. VEP studies together with detailed ophthalmologic examinations supply important information regarding optic-nerve involvement in GBS. However, the clinical correlation between optic-nerve involvement and the prognosis is unclear, and remains an area for future investigation.


 XML Download
XML Download