

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Hypohidrotic ectodermal dysplasia (HED) is a genetic disorder characterized by defective development of teeth, hair, nails, and eccrine sweat glands. The most prevalent form of HED is inherited as an X linked condition; however, autosomal dominant and autosomal recessive forms of the disorder have been described1. Clinical features of HEDs include sparse and fine hair, missing or conical-shaped teeth, decreased sweat and mucous glands, hypoplastic skin, and heat intolerance with exercise or increased ambient temperature.
Glucose-6-phosphate dehydrogenase (G-6-PD) is the first enzyme in the pentose phosphate pathway of glucose metabolism and its deficiency results in diminished reductive energy of red cells and may result in hemolysis. It is an X-linked recessive enzymatic defect and is fully expressed in hemizygous males and homozygous females2.
CASE REPORT
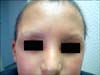
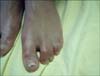
A 12-year-old boy presented to the outpatient clinic with the complaints of hair and eyebrow disturbances, teeth disfigurement, decreased sweating, and xerosis. Dermatological examination revealed sparse hair and eyebrows, conical-shaped teeth, xerosis, syndactylia, transverse grooves, and discoloration of the nails (Fig. 1, 2, 3). The general physical examination revealed no pathological finding and no problems with hearing or sight were detected. His 3-year-old sister also had sparse hair and eyebrows, xerosis, and syndactylia. Her teeth were normal. In both cases, decrased sweating was demonstrated with an iodine-starch sweat test (An iodine solution is applied to sweaty areas. After drying, starch is sprinkled on the area. If the starch-iodine combination turns a dark blue color, it is concluded that there is excess sweat). His 11-year-old sister and parents were clinically normal. His parents were not relatives and there was no important disease in his family history. Examination of routine laboratory findings found no problem, except for anemia. His parents confirmed that he had severe and prolonged neonatal jaundice at birth; after that, an exchange transfusion was performed. G-6-PD enzyme assay was performed for differential diagnosis of hyperbilirubinemia and hemolytic anemia. Diagnosis of G6PD deficiency was confirmed by a spectrophotometric method. His G-6-PD enzymatic activity level was low. G-6-PD enzymatic activities of his sisters were in normal ranges.
DISCUSSION
HED was first described by Thurnam in 1848 and later by Darwin in the 19th century. It was assigned to the X chromosome in 1921 by Thadani, who later reported that female carriers could manifest signs of the condition3. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is one of the more common types of over 150 clinically distinct hereditary ectodermal dysplasias. It is characterized by sparse hair, abnormal or missing teeth, and inability to sweat due to lack of sweat glands4. Affected persons show distinctive facial features, with frontal bossing, depressed nasal bridge with a saddle nose, and large lower lips. Corneal and lenticular opacities or conductive hearing loss are uncommon features. However, absence of lacrimal puncta is a characteristic finding5. HED is caused by defects in the ectodysplasin signal transduction pathway. Mutations in the gene encoding the ligand ectodysplasin A (EDA) underlie classic, X-linked recessive HED, whereas mutations in genes encoding the EDA receptor and the adaptor protein associated with the EDA receptor's death domain result in autosomal dominant and autosomal recessive forms of HED6. Treatment of HED is supportive and includes protection from heat exposure, early denture fittings, skin, hair, ear, nose, and nail care, and genetic counseling for family planning7.
G6PD deficiency, an X-linked recessive enzymatic defect in the hexose monophosphate shunt protecting against cellular damage from oxidative stress, is a common hematological problem worldwide8. G-6-PD is expressed in males carrying a variant gene, while heterozygous females are usually clinically normal9. G-6-PD-deficient individuals are usually asymptomatic; however, in some cases, exposure to chemicals and drugs can induce massive intravascular hemolysis. Major findings of this enzymatic deficiency include jaundice, and acute hemolytic and chronic nonspherocytic hemolytic anemia10.
In this report, our patient had many symptoms (sparse hair and eyebrows, conical-shaped teeth, xerosis, transverse grooves, and discoloration of nails) of HED. He was clinically normal; however, his past medical history revealed a diagnosis of G-6-PD deficiency after investigation of prolonged neonatal jaundice. His 3-year-old sister also had sparse hair and eyebrows, xerosis, and syndactylia, while the other sister showed no signs of HED, and their G-6-PD enzymatic activities were in normal ranges.
According to one report, the locus for G6PD has been assigned to the region distal to Xq2611 and the AED locus was on Xq12-q13.15. It has been postulated that the AED locus is nearer to the X chromosome centromere than G6PD11. Although it has been mentioned that loci of ectodermal dysplasia and G-6-PD deficiency genes are near to one another, we did not find any case study reporting on occurrence of these two X-linked genetic diseases together in the literature. To the best of our knowledge, we present here the first documented case of HED associated with G-6-PD deficiency.
With the aspect of this rare and interesting case, the relationship between HED and G-6-PD deficiency has been discussed in this manuscript.

 XML Download
XML Download