

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Tracheobronchopathia osteochondroplastica (TO) is a rare disorder of unknown cause that affects the tracheobronchial tree. It is characterized by the presence of multiple osseous and/or cartilaginous submucosal nodules protruding into the lumen of the trachea and large bronchi, which typically spares the posterior wall.1,2 Amyloidosis is an uncommon disease, in which various organs are infiltrated by amorphous extracellular eosinophilic material, composed of insoluble proteins that have a high degree of beta-pleated sheet protein structure.3 TO can especially show asthma like symptoms.1
We describe a case that was initially thought to be asthma bronchiole but was later diagnosed as TO and tracheobronchial amyloidosis.
CASE REPORT
A 53-year-old man had a history of chronic cough and wheezing, recurrent lower respiratory tract infections, and progressive shortness of breath. Asthma had been misdiagnosed in the previous 3 years of this patient, and his therapy included an inhaled beta-adrenergic agonist and glucocorticoid. He denied coughing, stridor, and hemoptysis. His medical history was not significant for tobacco abuse.
On admission, his temperature was 36.9℃, pulse rate was 89 beats/min, respiratory rate was 19 breaths/min, and BP was 135/85 mm Hg. Auscultation of his lungs revealed diffuse rhonchi, and palpation revealed no abdominal organomegaly. The results from the examination of the centural nervous system (CNS) and skin were normal. Furthermore, physical examination of oral cavity and oropharynx was normal.
The results of his blood, chemistry, liver function, urine tests including creatinine clearance rate, urinalysis for urinary protein, beta-2 microglobulin, and serum immunoglobulin levels, renal ultrasonography, echocardiography, and electrocardiography were normal. Assays for human immunodeficiency virus and antineutrophil cytoplasmic antibodies were negative, and his rapid plasma reagin was nonreactive. Pulmonary function tests were indicative of an obstructive airway disease [Forced Vital Capacity (FVC): 49% of predicted, Forced Expiratory Volume (FEV) 1: 43% of predicted and FEV1/FVC: 68% of predicted], and the reversibility test was found to be negative.
The chest radiograph at admission showed homogenous opacity in the right middle lobe which is consistent with the symptoms of atelectasia.
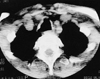
Computed tomography showed the prominent cartilaginous nodules protruding into the airway lumen. Other roentgenologic findings include the thickening or irregularity of the tracheal and/or bronchial walls with sparing of the membranous posterior wall, deformed tracheal cartilage rings without evidence of external compression or submucosal calcification, and atelectasis of the right middle lobe (Fig. 1).
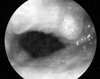
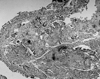
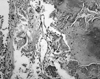
Fiberoptic bronchoscopic examination revealed mucosal nodules protruding into the lumen of the trachea (Fig. 2). These lesions extended throughout the trachea but extended especially in the middle and lower thirds; nodules were also present in the carina and in the medial wall of the two main bronchi. The posterior walls of the trachea and main bronchi were characteristically not involved. The lumen of the right middle lobe bronchus was completely obstructed. The initial diagnosis was tracheopathia osteoplastica based only on visual inspection through bronchoscopy. Multiple biopsies from the mucosal nodules demonstrated hyalinised, degenerated nodules of cartilage that were associated with fibrous tissue, spotty calcifications and homogenous, eosinofilic nodules in the submucosa (Fig. 3). However, amyloidosis was added to the diagnosis because characteristic findings of amyloid were obtained in tissue specimens stained by hematoxylin-eosin and congo red dye, and these specimens exhibited green birefringence under polarized microscopy (Figs. 4 and 5).
The patient underwent extensive testing in order to rule out systemic amyloidosis. Fat pad aspirates, a highly sensitive assay for systemic amyloid deposition, did not bind to the congo red stain. Serum or urine protein electrophoreses failed to detect any monoclonal protein. Bone marrow biopsy and aspirate did not identify plasmacytosis. The case did not exhibit clonal expansion of the kappa or lambda light chain immunoglobulin in the bone marrow through immunohistochemical staining. Resting electrocardiograms or echocardiograms did not manifest any signs of infiltrating cardiac disease.
DISCUSSION
TO is a rare, benign condition and is characterized by the presence of bony and cartilaginous nodules in the tracheal and bronchial mucosa.4 The entity was named "tracheopathia osteoplastica" in 1910 by Aschoff.1 Secondary systemic amyloidosis (amyloid A chain; AA type) is usually secondary to neoplastic or chronic inflammatory processes, including multiple myeloma, rheumatoid arthritis, bronchiectasis, tuberculosis, etc.5 The current consensus is that TO and tracheobronchial amyloidosis are not related because each has its distinct pathology. TO does not exhibit positive congo red staining, and tracheobronchial amyloidosis is not associated with bony or cartilaginous formation. The confusion between these two disease processes has arisen because of similarities in their bronchoscopic appearance.6
Localized pulmonary amyloidosis, in which amyloid deposition is restricted to the respiratory system, is a rare disease and occurs in three histopathologic types: tracheobronchial, nodular, and diffuse parenchymal.5 However, respiratory tract involvement has not been a clinical feature in over 200 patients with systemic AA amyloidosis who were evaluated.7 Since AA amyloidosis is a generalized disease, it may be diagnosed in any amyloid-containing tissue, but the most common biopsy sites are subcutaneous fat, kidney, rectum, and liver. When amyloid is present in small amounts, it can be easily overlooked.8 In our case, AA amyloidosis was confirmed histopathologically but amyloidosis was not discovered in other organs except in the lung tissue. The combination of TO and amyloidosis is rarely reported. Leske et al.1 reported 16 biopsy specimens that were examined specifically for amyloidosis after congo red staining, and only 2 of 16 specimens (13%) disclosed both TO and amyloidosis. Therefore, amyloidosis should be pathologically excluded in patients thought to have TO.
Bronchoscopy remains the best technique in diagnosing TO. Bronchoscopy characteristically reveals multiple isolated or confluent-appearing, submucosal, bony and cartilaginous nodules measuring 1 to 6 mm in diameter. The TO lesions are dispersed more commonly in the distal two thirds of the trachea. The nodules are projected into the airway lumen at variable depths. The involvement of the lobar and more peripheral bronchial tree is very rare.1 In bronchoscopy, hyperemic mucosal nodular lesions were detected instead of submucosal bony and cartilaginous nodules. These nodules completely obstructed the right middle lobe, but the involvement of only the anterior and lateral walls of the pulmonary system suggested TO. It has been reported that bronchoscopic appearance alone is sufficient enough to establish the diagnosis of TO; imaging and biopsy specimens are usually unnecessary. However, in cases with mucosal nodular involvement and obstruction of the lobe bronchus by nodules, biopsy should be taken to exclude amyloidosis. In contrast to TO, tracheobronchial amyloidosis typically does not spare the posterior membranous wall. In the rare cases of TO that involves also the posterior walls, amyloidosis may be excluded by the pathologist using congo red dye in search of the typical green birefringence under polarized light microscopy.1 In our case, posterior trachea and bronchi wall involvement was not detected. As it is presented in our case, amyloidosis can sometimes show involvement of only the anterior and lateral walls, which confirms that diagnosis biopsy may be needed.
Patients with tracheobronchial amyloidosis have symptoms that are similar to those caused by various airway disorders.6 TO that simulates asthma has been described previously and should be included in the differential diagnosis of large airway disorders mimicking asthma.1 Some authors have found an association between TO and bronchial asthma.9,10 Vivian et al. reported that 20% of their cases had a history of asthma-like symptoms, and 32% whoed signs of wheezing at auscultation.1 In our case we also found asthmatic symptoms that lasted for 3 years. TO and amyloidosis should also be considered in elderly asthmatic patients who are unable to be treated.
The management of patients who were diagnosed antemortem with TO is typically conservative and symptomatic because most patients do not experience serious morbidity from the disorder.4 The management of tracheobronchial amyloidosis is also largely dependent upon dyspnea which is related to bronchial obstruction and may involve intermittent bronchoscopic resection, surgical resection, carbon dioxide laser ablation, and Nd: yttrium aluminum garnet (YAG) aser therapy. Repeated bronchoscopic intervention is thought to be preferable and safer than open surgery. Stenting may have a role for narrowing of the airways and complications thereof.7,11 No progression of the disease has been detected in the patient after being followed up for two years.

 XML Download
XML Download